Water matters: Enhancing early drug discovery with insights from water energetics
Share
In the race to develop breakthrough therapeutics, understanding the role of water energetics can contribute novel insights to unlock new design strategies. Water molecules in and around protein binding sites are not just passive bystanders—they play an active role in molecular recognition and can influence ligand binding affinity, selectivity, and stability. Incorporating explicit water energetics into your hit discovery, docking, and lead optimization workflows provides a more nuanced view of protein-ligand interactions, helping you to predict binding outcomes more accurately and identify new opportunities to optimize leads.
This webinar will discuss the impact of two technologies that leverage explicit water energetics in the binding pocket to enhance drug design—WaterMap and Glide WS. Through evaluation of the thermodynamics of water sites, WaterMap targets high-energy, unstable hydration sites to improve the identification of better hits and more potent leads. Glide WS is a molecular docking tool that uses detailed water analysis from WaterMap calculations to evaluate the impact of desolvation events on protein-ligand binding. The combination of WaterMap and Glide WS empowers accurate pose prediction and enhances early enrichment in modern virtual screens.
We will demonstrate the impact of water energetics through Schrödinger’s collaborative SARS-CoV-2 project that led to the rapid discovery of lead compounds with unique SAR that exhibited potent enzymatic and cellular activity with excellent pan-coronavirus coverage.
Webinar Highlights
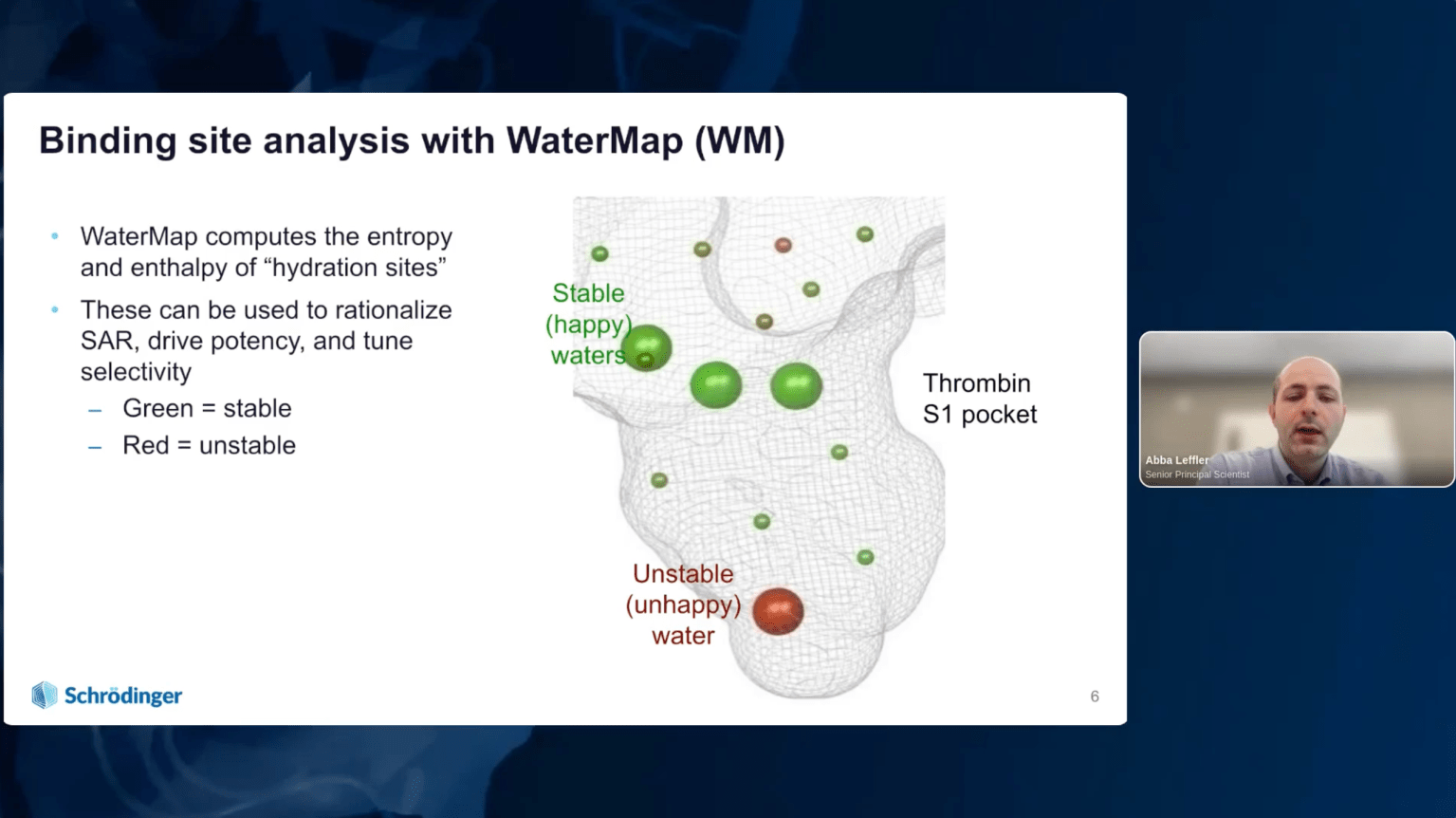
Overview of the role of water molecules in protein binding sites and their impact on ligand docking outcomes
How modeling explicit water energetics can uncover new binding modes and reduce false positives
Lead optimization insights using hydration patterns to boost selectivity and drug-like properties
Success stories from Schrödinger’s drug discovery projects
Our Speakers
Abba Leffler
Senior Principal Scientist, Therapeutics Group, Schrödinger
Abba E. Leffler, Ph.D. is a Senior Principal Scientist in the therapeutics group at Schrödinger, where he currently focuses on small-molecule drug discovery. He received his AB in Chemistry with a Certificate in Applied Mathematics from Princeton University, after which he worked at D. E. Shaw Research before going on to obtain his PhD in Neuroscience from NYU School of Medicine. His research has been published in Science, The Journal of Neuroscience, The Journal of Chemical Information and Modeling, and Proceedings of the National Academy of Sciences among others. He is an inventor on multiple patents as well.
Gary Zhang, Ph.D. is the product manager of Schrödinger docking technologies. Gary leads the team improving the performance and expanding the domain of applicability of Schrödinger docking tools. He received his Ph.D. from Duke University working on engineering charge transfer pathways in biological systems and followed by postdoctoral training at Scripps Research working on improving peptide docking performance.
Modeling lipid nanoparticles: Self-assembly and apparent pKa calculation
Share
Abstract:
The application of lipid nanoparticles (LNPs) for the delivery of nucleic acids to address various health conditions is an active area of research and development. However, the limited understanding of the relationships between composition, structure, and performance hinders innovation, mainly due to the limited structural information that can be easily determined by experiment.
In this webinar, we will introduce Schrödinger’s coarse-grained simulation technology that can simulate the formation of LNP structures from a random mixture. This model enables structure-based design of LNPs by correctly reproducing many known structural features and some key behaviors (e.g. bleb formation as a function of pH) of LNP formulations. Another key aspect of LNP formulation is the design or selection of ionizable lipids (ILs) with appropriate apparent pKa values. We will present a structure-based, atomistic simulation technology for calculating formulation-specific apparent IL pKa values, which is designed to elucidate the factors which influence the apparent pKa and aid in IL selection.
Webinar Highlights:
Simulation of the formation of LNP structures using coarse-grained molecular dynamics (MD) modeling technology
Calculation of apparent pKa values of ILs via a structure-based, computational methodology
Overview of Schrödinger’s computational solutions for understanding self-assembly and structural properties of LNPs
Our Speaker
John Shelley
Fellow, Schrödinger
John earned a MSc from the University of Waterloo in theoretical chemistry and a PhD from the University of Pennsylvania in computational chemistry. Following post-doctoral research in computational chemistry at the University of British Columbia, he worked for Procter & Gamble studying surfactant structures in solution. For the last 23 years, John has worked for Schrödinger, LLC, as a scientific software developer and a research scientist, managing a number of products including the Materials Science Coarse-Grained product. John has focused on computer modeling of drug formulations for much of the last 8 years.
【ポスター討論 1P-17】
日時:11月20日(水) 17:30 – 19:00
演題 : In Silico Enabled Discovery of KAI-11101, a Preclinical DLK Inhibitor for the Treatment of Neurodegenerative Disease and Neuronal Injury
井川 英之(Therapeutic Group, Schrödinger)
【ランチョンセミナー LS-5】
日時 : 11月21日(木) 12:20-13:40
演題 : The Predict-First Paradigm: How Digital Chemistry is Shaping the Future of Drug Discovery
井川 英之(Therapeutic Group, Schrödinger)
Schrödinger is excited to be participating in the 2024 Advanced Automotive Battery Conference taking place on December 9th – 12th in Las Vegas, Nevada. Stop by booth #908 to speak with us.
Schrödinger is excited to be participating in the MRS Fall 2024 conference taking place on December 1st – 6th in Boston, Massachusetts. Join us for presentations by Schrödinger scientists on Dec 4th and 5th. Additionally, attend a presentation on Dec 3rd by Panasonic, co-authored by Schrödinger, titled “Discovering Low-Viscosity Molecules Using an Integrated Physics-Based Modeling, High-Throughput Screening, and Active Learning Approach (2)— Screening from PubChem Database.”
Discovering Low-Viscosity Molecules Using an Integrated Physics-Based Modeling, High-Throughput Screening, and Active Learning Approach (2)— Screening from PubChem Database
Presenters: Nobuyuki Matsuzawa, Hiroyuki Maeshima, Tatsuhito Ando, Atif Afzal, Benjamin Coscia, Andrea Browning, Mathew Halls, Karl Leswing, Tsuguo Morisato
Schrödinger collaborated with Panasonic on this presentation
DEC 4 | 3:45 PM
Hynes, Level 3, Ballroom C
Discovering Low-Viscosity Molecules Using an Integrated Physics-Based Modeling, High-Throughput Screening and Active Learning Approach (1)— Screening from the GDB Database
Speaker: Atif Afzal, Principal Scientist
Abstract:
The discovery of low-viscosity molecules is crucial for the development of next-generation batteries and capacitors. Large molecular libraries available in the literature provide a valuable resource for identifying promising candidates. In this study, we utilized the GDB database1, one of the largest repositories of small molecules, to identify low-viscosity molecules. We employed and benchmarked molecular dynamics methods to accurately compute the dynamic properties without the need for synthesis or empirical testing, validating our calculations against experimental data. However, the number of molecules of interest from the GDB database is too large (several hundreds of thousands), making it impractical to identify promising candidates using purely physics-based models due to computational costs. Therefore, we implemented advanced machine learning (ML) techniques and smart selection approaches to dramatically reduce the number of physics-based calculations needed. Physics-based simulations of viscosity included both Green-Kubo and Einstein-Helfand approaches allowing for robust calculation across the selected molecules. By employing an active learning approach, we optimized the selection of molecules, enhancing the efficiency of the ML model while targeting low-viscosity candidates. Additionally, we computed the boiling points (BP) of the molecules using ML models trained on experimental BP data. As a result, we identified more than 100 molecules with viscosities less than 0.35 cP and BP above 80°C. We demonstrate that by integrating accurate physics-based models with advanced ML techniques, we can effectively identify top molecular candidates while significantly reducing computational costs.
DEC 5 | 11:15 AM
Sheraton, Second Floor, Constitution B
Prediction of aqueous and non-aqueous solubility using machine learning
Speaker: Lihua Chen, Senior Scientist
Abstract:
Solubility, the capacity of a solute to dissolve in a solvent, forming a solution, is a crucial design parameter across various materials and life science applications. Due to the high cost of experimental measurements, we have developed quantitative structure-property relationship (QSPR) models to rapidly and accurately predict aqueous solubility in water and non-aqueous solubility in organic solvents. For this purpose, we gathered 14,485 room temperature aqueous solubility data points and 45,313 temperature-dependent non-aqueous solubility data points from literature and open-source databases. Additionally, we incorporated advanced cheminformatics-based, graph-based, and physics-based descriptors computed through classical molecular dynamics to optimize machine learning performance. These models can significantly streamline molecular discovery by providing rapid, accurate solubility predictions, reducing the need for costly experiments, and accelerating the identification and optimization of promising candidates.
Glide has long set the gold standard for commercial molecular docking software due to its robust performance in both binding mode prediction and empirical scoring tasks, ease of use, and tight integration with Schrödinger’s Maestro interface and molecular discovery workflows.
To mark the 20th anniversary of the original publication of Glide in 2004, one of the most cited papers in the Journal of Medicinal Chemistry to date1, Schrödinger is proud to introduce Glide WS, a new molecular docking workflow within Glide which leverages water energetics from WaterMap. The release of Glide WS further demonstrates Schrödinger’s commitment to the development of accurate tools that meet the needs of today’s drug discovery teams.
Glide WS offers:
Enhanced pose predictions by incorporating a flexible description of explicit water molecules
Efficient scoring and prioritizing of ligands to improve hit enrichment rates
Reduced false positives by filtering out compounds that are incompatible with the target binding site in virtual screens
A comprehensive scoring function calibrated by FEP+ calculations and experimental data
Key Features
Glide WS is a molecular docking workflow built on the foundation of Glide SP2, 3 and WScore4 that provides significantly improved sampling and scoring of small molecules in the binding pocket. The Glide WS scoring function was developed with quantitative guidance from experimental data and FEP+ calculations. It uses information from WaterMap5 to evaluate the water energetics associated with desolvation and capture water mediated interactions. Additionally, Glide WS incorporates MM-GBSA calculations to assess the overall goodness of fit of the ligand pose in the binding site.
Enhanced conformation generation for pose prediction
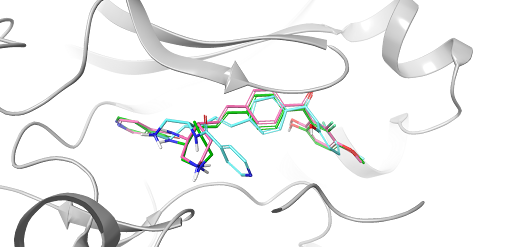
Glide WS computes the ligand conformer distribution using a novel approach that combines an RDKit algorithm and ConfGen. By focusing on ‘hard to sample’ ligand features such as non-aromatic rings, this new method realizes substantial gains in the generation of native-like ligand conformers while keeping computational costs within reach for most practical docking and virtual screening applications. As shown in Figure 1, improved ring sampling in Glide WS predicted the correct conformation of the 7-member ring in the middle of the molecule.
Figure 1: Redocking protein kinase A (1SVH) with Glide WS (pink pose) showed improved pose prediction compared to Glide SP (blue pose). The crystal pose is shown in green.
Scoring function calibration guided by FEP+
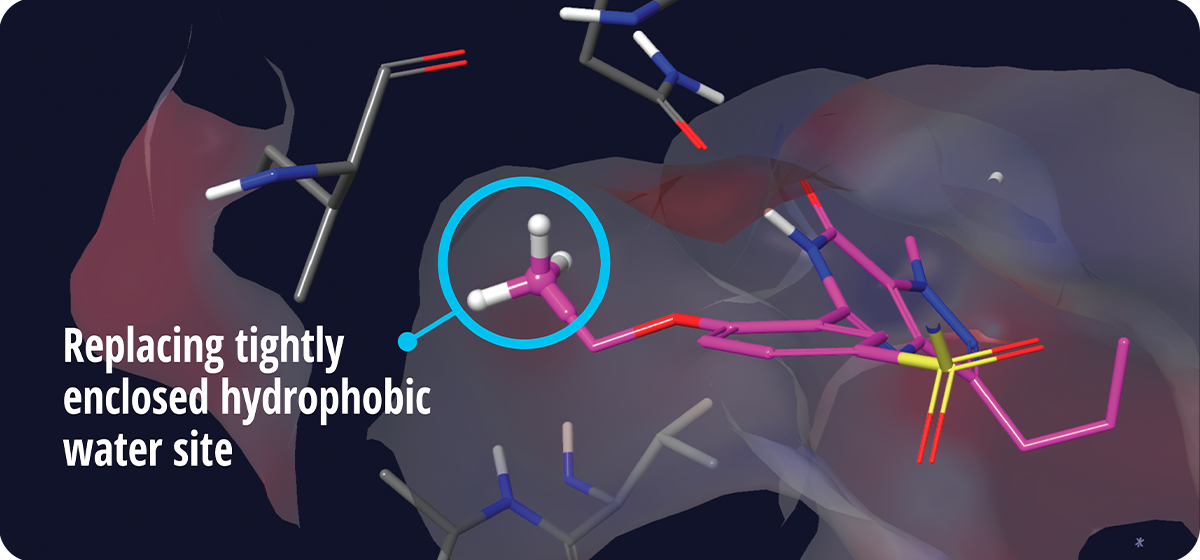
The Glide WS scoring function is calibrated by FEP+6, a widely established benchmark computational method for binding affinity prediction, to yield more comprehensive reward and penalty terms. The scoring function in Glide WS is more robust than Glide SP, with many additional scoring terms based on a detailed study of thousands of PDB structures. As an example, Glide WS can identify “magic methyl” sites in good agreement with FEP+ and experimental data, wherein the addition of a single heavy atom can provide a seemingly nonintuitive boost in the potency of the ligand (Figure 2). By accurately capturing this important effect, Glide WS is able to achieve better separation of true ligands from false positives across many targets.
Superior performance on self-docking
In a curated protein-ligand dataset with 765 PDB complexes, Glide WS is able to reproduce the ligand pose for 98% of the cases compared to 88.7% for Glide SP and 91% for Glide XP7 (Table 1). However, Glide WS is notably slower than Glide SP due to the extensive sampling and more comprehensive scoring of the poses. Glide WS is best used to refine and filter a subset of compounds from Glide SP screening as discussed in the section below.
Table 1: Glide WS outperforms Glide SP and XP on a curated protein ligand complex dataset covering 61 drug targets.
Outperforms Glide SP on key virtual screening benchmark
A common use case of molecular docking is in silico screening of real and virtual compounds to identify novel hits. As the number of compounds experimentally tested will typically be much less than the number screened, it is vital that a docking method be able to recover true binders near the top of the docking output— this is known as early enrichment. We compared the early enrichment performance of Glide WS and Glide SP on a diverse subset of 23 targets from DUD-E. DUD-E is a popular benchmark designed to evaluate the performance of docking tools (Figure 3). For each target, DUD-E provides a set of diverse ligands and property-matched decoys.
As Glide WS uses the difference between experimental binding affinity and docking score of the native ligand to estimate reorganization energy, the Glide WS docking score is a more realistic estimation of the true binding affinities across different targets. Here, we applied a cutoff based on the Glide WS docking score (-8kcal/mol) and selected up to 100 ligands to mimic a real virtual screening campaign and considered the same number of ligands screened by Glide SP to have a fair comparison.
Figure 3: Protein family distribution of the DUD-E subset investigated.
Figure 4: (A) Glide WS and Glide SP show good early recovery of known active compounds. (B) Glide WS has fewer decoys with bad ABFEP scores than Glide SP among top ranked ligands.
Both Glide WS and SP achieve reasonable hit rates for the targets investigated, with Glide WS finding more active compounds as shown in Figure 4a. This type of analysis does not consider the potential for the competing decoys to be active and potentially underestimates the value of rescoring top hits.
We further investigate the decoys using absolute binding free energy perturbation (ABFEP)8. ABFEP provides a theoretically more rigorous and accurate description of protein–ligand binding thermodynamics, predicting the binding affinities with errors on the same order of magnitude of experimental variations. This investigation helps us better understand the nature of the high-ranking decoys and identify clear false positives coming out from the Glide WS and SP screens, i.e., decoys with good docking scores but bad ABFEP scores. As shown in Figure 4b, Glide WS generates much fewer bad decoys (ΔGABFEP+ greater than -7kcal/mol) than Glide SP among the top scored ligands. Thus, we find, based on the ABFEP scoring data that the Glide WS decoys are much more likely to be active compounds than the Glide SP decoys. This in turn substantiates that Glide WS is a more effective filtering tool in a virtual screening campaign for selecting compounds to be purchased or to be rescored with ABFEP within the context of a full virtual screening funnel.
When to use Glide WS
Glide WS is a more advanced docking tool than Glide SP, providing better poses and fewer false positives, while still being much less expensive than ABFEP+. As Glide WS is ~20x slower than Glide SP, we recommend using Glide WS to further filter virtual hits from a Glide SP screening. Glide WS can also be used in a lead optimization stage to help filter ideas coming from enumeration or other ideation tools.
Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field.