Designer Fluorescent Redoxmer Self-Reports Side Reactions in Nonaqueous Redox Flow Batteries
Gaining molecular insights towards inhibition of foodborne fungi Aspergillus fumigatus by a food colourant violacein via computational approach
Uncovering the light absorption mechanism of the blue natural colorant allophycocyanin from Arthrospira platensis using molecular dynamics
Improving color and digestion resistibility of 3D-printed ready-to-eat starch gels using anthocyanins
Molecular insights into the structure forming properties of zein and a rheological comparison with hordein
Steviol rebaudiosides bind to four different sites of the human sweet taste receptor (T1R2/T1R3) complex explaining confusing experiments
Advancing the design and optimization of drug formulations with combined computational and experimental approaches

Advancing the design and optimization of drug formulations with combined computational and experimental approaches
Scientists from AbbVie and Schrödinger collaborated to systematically investigate amorphous solid dispersion (ASD) dissolution behaviors by combining thermodynamic modeling, molecular simulation, and experimental research.
Summary
Investigated ASD/water interfacial gel layer behavior with thermodynamic modeling and confirmed this behavior by microscopic erosion time test (METT) experiments
Acquired additional insights of ASD dissolution at the molecular level, which validated the results from thermodynamic modeling and METT experiments, and inspired the design of drug formulations
Gained a deeper understanding of the drug load-dependent release mechanism and the occurrence of loss of release of a complex ASD formulation that is comparable to commercially available formulations

Challenges
During the dissolution of amorphous solid dispersion (ASD) formulations, the drug load (DL) often impacts the release mechanism and the occurrence of loss of release (LoR). The effect has been experimentally observed and discussed in the literature. However, the underpinning principles and mechanisms are less investigated and reported, since they require a better understanding of the thermodynamics and molecular interactions of ASD dissolution. A combined approach leveraging molecular-level simulations and thermodynamic modeling is uncommon in the literature, but can provide deeper insights into the complex phase behavior of ASDs during dissolution.
Approach
Continuing their collaboration on ASD dissolution R&D,1-2 scientists from AbbVie and Schrödinger extended this research to use a combined experimental-computational approach to investigate the drug load-dependent release mechanism and the occurrence of loss of release in ASDs containing the drug ritonavir and the polymer copovidone (PVPVA64). All molecular simulations were performed using Schrödinger’s Materials Science platform (MS Maestro) and Desmond for molecular dynamics (MD) and coarsegrained (CG) simulations.
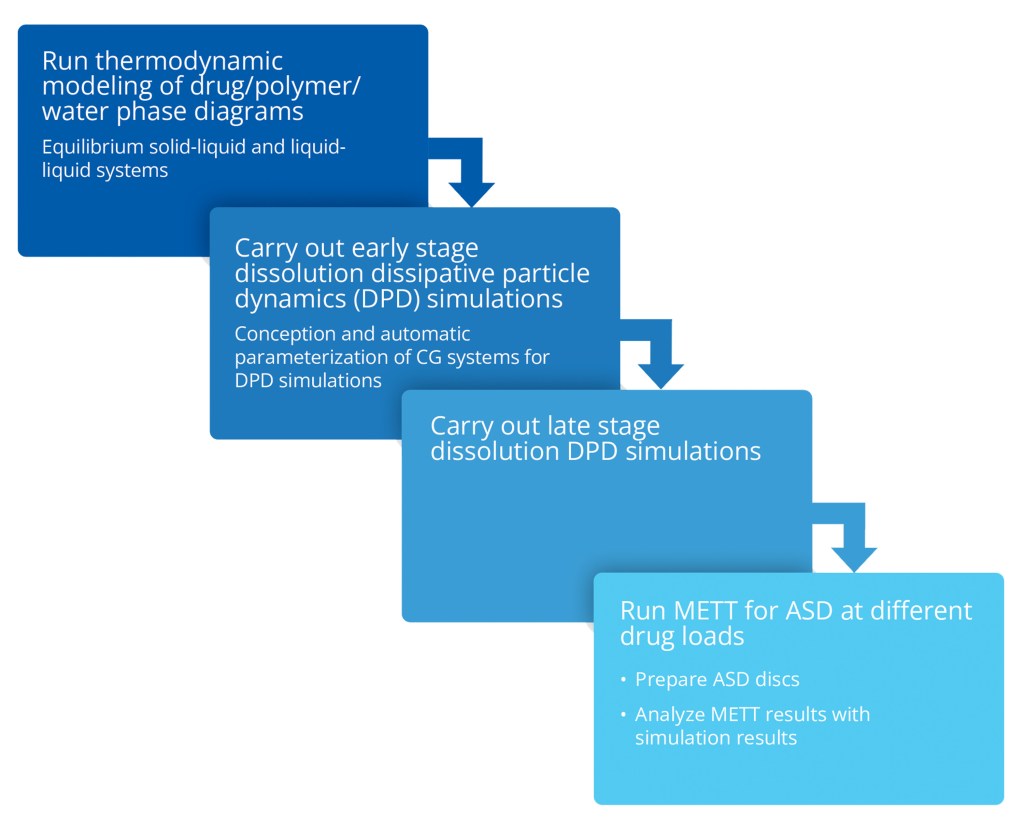
Results and outlook
This study3 demonstrates that the complementary information gained from thermodynamic modeling, molecular simulation, and experiment provides a valuable and more complete understanding of the release mechanism of ritonavir/PVPVA64-based ASDs.
- At low to moderate DL (5–15 wt%), PVPVA64 formed the major phase (polymer-rich phase), while ritonavir formed the minor phase (drug-rich phase) and the drug release is dictated by its solubilization. At high DL (20–40 wt%), self- and drug-polymer interactions play a larger role.
- For high DL (40 wt%) ASDs, at the early stages of hydration, the drug molecules distinctively aggregate into drug-rich phase clusters near the surface of the ASD, delaying erosion and drug release considerably.
- In the late dissolution simulations, the interactions between the ritonavir molecules and the polymer’s monomers, as well as with the water molecules, were tracked to better understand the mutual association between drug, polymer, and solvent.
- Interaction analysis for late stage simulations focusing on the drug-rich clusters revealed how dehydration and stabilization of these clusters increases with high DLs.
This approach can support the identification of maximal DL as a possible starting point for further ASD formulation design and development. The predictive power of the presented models provides scientists with powerful tools to overcome challenges in optimizing drug formulations.
Methodology implemented for molecular simulation component of the investigation
Automatic parameterization of coarsegrained (DPD) systems
Seamless simulation studies of complex formulations
Description of complex dissolution/ organization patterns
References
-
Case study
Advancing the design and optimization of drug formulations with coarse-grained molecular simulations
-
Molecular-Level Examination of Amorphous Solid Dispersion Dissolution
Mohammad Atif Faiz Afzal, Kristin Lehmkemper, Ekaterina Sobich, Thomas F. Hughes, David J. Giesen, Teng Zhang, Caroline M. Krauter, Paul Winget, Matthias Degenhardt, Samuel O. Kyeremateng, Andrea R. Browning, and John C. Shelley Mol. Pharmaceutics 2021, 18, 11, 3999-4014
-
Predicting the Release Mechanism of Amorphous Solid Dispersions: A Combination of Thermodynamic Modeling and In Silico Molecular Simulation
Stefanie Walter, Paulo G. M. Mileo, Mohammad Atif Faiz Afzal, Samuel O. Kyeremateng, Matthias Degenhardt, Andrea R. Browning and John C. Shelley Pharmaceutics 2024, 16(10), 1292
LOPEC 2025

Conference
LOPEC 2025
- February 25th-27th, 2025
- Munich, Germany
Schrödinger is excited to be participating in the LOPEC 2025 taking place on February 25th – 27th in Munich, Germany. Join us for a presentation by Hadi Abroshan, Principal Scientist at Schrödinger, titled “Integrating Atomistic Simulations, Machine Learning, and Cloud-Based Collaboration for Next-Generation Electronic Materials.” Stop by booth #B0313 to speak with Schrödinger scientists.
Click here to learn how Schrödinger’s digital chemistry platform empowers you to discover novel optoelectronics materials.
Integrating Atomistic Simulations, Machine Learning, and Cloud-Based Collaboration for Next-Generation Electronic Materials
Speaker:
Hadi Abroshan, Principal Scientist, Schrödinger
Abstract:
The creation of next-generation display technologies hinges on innovative research strategies and collaborative tools. This presentation highlights how the integration of physics-based simulations, machine learning (ML), and a cloud-enabled platform accelerates the discovery and refinement of advanced optoelectronic materials.
We introduce the Schrödinger digital chemistry platform, which facilitates advanced simulations of optoelectronic materials, spanning from individual molecules to thin films developed via vacuum deposition or solution processing. This platform’s automated capabilities predict key material properties such as electronic transitions, hyperfluorescence, charge carrier mobility, refractive index, thermophysics, interfacial mixing, and molecular orientation.
We then explore the synergy between physics-based simulations and ML, which significantly streamlines the materials discovery process. By analyzing large datasets and identifying trends, ML allows for faster predictions of material properties. An active learning screening process efficiently pinpoints promising candidates based on multiple properties, all while reducing computational cost.
Further, we discuss genetic optimization algorithms that drive the development of new materials for electroluminescent devices. These algorithms emulate natural selection, refining material properties iteratively to uncover high-performance compounds tailored to specific targets. When combined with high-throughput screening, this approach accelerates the exploration of chemical space, leading to rapid material advancement.
Lastly, we introduce Schrödinger’s LiveDesign, a web-based collaboration platform that enhances modern R&D by integrating advanced modeling, data management, and ideation. LiveDesign empowers research teams to collaborate effectively, regardless of geographic location, supporting a seamless end-to-end research workflow.
アーカイブ配信: E-sol to Predict Unbound Brain-to-Plasma Partition Coefficient, Kp,uu

Webinar
アーカイブ配信: E-sol to Predict Unbound Brain-to-Plasma Partition Coefficient, Kp,uu
- November 13th, 2024
- Virtual
水和エネルギー(E-sol)は、創薬において重要な脳内移行性予測の強力な指標です。
E-solは、非結合脳漿中分配係数(Kp,uu)と高い相関性を持ち、多様な化学構造やデータセットにおいて、従来法よりも正確な予測を実現します。
弊社のソフトウェアでは、E-solを用いることで、脳内移行性の高い化合物ライブラリ構築や、効率的な化合物スクリーニングが可能になります。
本ウェビナーでは、E-solの基本から応用事例、FAQまでを解説し、創薬研究におけるE-sol活用の可能性を探ります。
本ウェビナーは日本語での発表です。
このウェビナーの録画視聴をご希望の方は、こちらの申込書にご記入ください。
※ご質問、ご不明な点がございましたら下記までお問い合わせください。
シュレーディンガー株式会社 機能紹介ウェビナー事務局
E-mail: info-japan@schrodinger.com
The 3rd Annual Meeting of The Antibody Society of Japan

Conference
【12月9日(月)~12日(木)】『第3回日本抗体学会学術大会』ランチョンセミナー・出展
- December 9th-12th, 2024
- Sendai, Japan
シュレーディンガー株式会社は、第3回日本抗体学会学術大会に参加いたします。
会場: 仙台国際センター(宮城)
【ランチョンセミナー】
日時:12月11日(水) 12:30 – 13:30
演題: Schrödinger’s approach to physics-based antibody analysis and design
発表者: 市原収(シュレーディンガー株式会社)
【ブース No.26】
Schrödinger 創薬支援プラットフォームと受託解析サービスをご紹介します。
- 最新のドッキングシミュレーション解析
- 高速高精度な結合自由エネルギー予測
- 多機能なリガンドベース解析
- 機械学習を用いた解析
- 抗体構造モデリング
- タンパク質間相互作用解析
- 化合物のシミュレーションデータ、実験データの管理、共有、解析等ディスカッションプラットフォーム
- 各シミュレーションの受託解析サービス
SCC78 2024

Conference
SCC78 2024
- December 11th-13th, 2024
- Los Angeles, California
Schrödinger is excited to be participating in the SCC78 conference taking place on December 11th – 13th in Los Angeles, California. Join us for a presentation by Haidong Liu, Senior Scientist at Schrödinger, titled “Screening Antioxidant Ingredients Using Machine Learning and Physics-based Modeling .”
Screening Antioxidant Ingredients Using Machine Learning and Physics-based Modeling
Speaker:
Haidong Liu, Senior Scientist, Schrödinger
Abstract:
Antioxidants are an important ingredient for cosmetic products to alleviate oxidative stress. While high-throughput screening for new antioxidant candidates still remains challenging experimentally. And the data-driven machine learning models would require the input of a reliable dataset. Here we present an efficient computational approach that combines the physics-based and machine learning tools to address this issue, and this
approach only uses molecular structures as inputs.
We used molecular quantum mechanical (QM) calculation and machine learning to predict the antioxidant activity through hydrogen atom transfer (HAT) mechanism. We first constructed a library of flavonoid structures and then calculated the hydrogen dissociation energies of the hydroxyl group in solvents using QM. The machine learning model was trained and validated using the hydrogen dissociation energies from QM calculations. We can easily screen thousands of molecules, and this physics-based and machine learning combined approach can be used for other properties.