Understanding of complex spin up-conversion processes in charge-transfer-type organic molecules
Tuning the Mobility of Indacenodithiophene-Based Conjugated Polymers via Coplanar Backbone Engineering
Predicting the Release Mechanism of Amorphous Solid Dispersions: A Combination of Thermodynamic Modeling and In Silico Molecular Simulation
Insights into the binding mechanism of 2,5-substituted 4-pyrone derivatives as therapeutic agents for fused dimeric interactions: A computational study using QTAIM, dynamics and docking simulations of protein–ligand complexes
Coarse-Grained Simulation of mRNA-Loaded Lipid Nanoparticle Self-Assembly
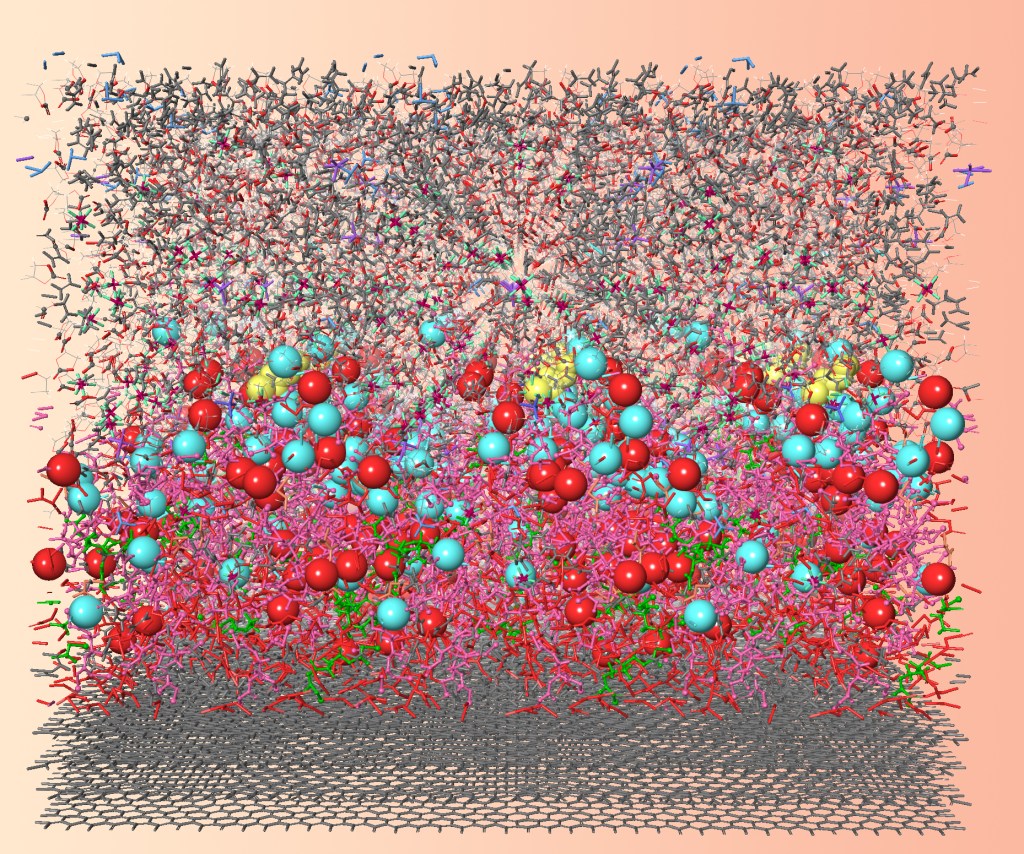
Towards long-life 500 Wh kg−1 lithium metal pouch cells via compact ion-pair aggregate electrolytes
Schrödinger Software 2024-3 新機能紹介ウェビナーアーカイブ配信

Webinar
Schrödinger Software 2024-3 新機能紹介ウェビナー アーカイブ配信
- October 15th, 2024
- Virtual
この度、最新版となる2024-3をリリースいたしました。
本ウェビナーでは、主要な新機能についてご紹介いたします。
このウェビナーの録画視聴をご希望の方は、こちらの申込書にご記入ください。
【ご紹介内容】
- Full release of the OPLS5 polarizable small molecule force field: Provides broad coverage of organic functional groups for improved FEP+ and Desmond simulation accuracy
- New Lambda Dynamics-enhanced protein residue mutation FEP+ to rapidly identify high quality protein variants (Beta): Enables a high-throughput computational workflow for protein design
- New Unbinding Kinetics workflow in Desmond for gaining insights into drug-target residence time (Beta): Applications to optimize in vivo efficacy, safety profiles, and ADMET
- Expanded DNA/RNA modeling toolkit: Improved accuracy in identification of RNA binding sites and new ability to interactively analyze protein-DNA/RNA interactions
※ご質問、ご不明な点がございましたら下記までお問い合わせください。
シュレーディンガー株式会社 ショートセミナー事務局
E-mail: info-japan@schrodinger.com
Machine learning for data-driven design of high-safety lithium metal anode
Computational and Machine Learning-Assisted Discovery and Experimental Validation of Conjugated Sulfonamide Cathodes for Lithium-Ion Batteries
Molecular-level insight into solubility-enhancement via cosolvents and amorphous solid dispersions


OCT 9, 2024
Molecular-level insight into solubility-enhancement via cosolvents and amorphous solid dispersions
Abstract:
A key to the viability of the newest generation of active pharmaceutical ingredient (API) candidates, which trend towards higher lipophilicity and lower water solubility, are the formulations engineered for their delivery. Although heuristics exist which help to identify effective delivery methods and choice of accompanying excipients, the novelty of each new drug frequently comes with unexpected formulation challenges. In this talk, we will highlight how molecular models can aid our ability to anticipate these challenges prior to candidate selection as well as to quickly understand issues that arise in later stages of development. We will discuss how these solutions can be executed by modelers or experimentalists alike in using the Schrödinger Materials Science platform.
Webinar Highlights:
- Prediction of solubility enhancement via organic cosolvents, using free energy perturbation (FEP+)
- Investigation of API dispersion in excipient and release in the gastrointestinal tract using all-atom and coarse-grained molecular dynamics simulations (MS CG and Desmond)
- Introduction to Schrödinger’s Materials Science platform for drug formulation modelers and experimentalists
Our Speaker

Ben Coscia
Principal Scientist, Schrödinger
Ben Coscia is a Principal Scientist at Schrödinger specializing in all-atom and coarse-grained molecular simulation of complex systems including polymers, soft matter and pharmaceutical formulations. Ben received his B.S. in chemical engineering from the University of Connecticut and then worked as a formulation scientist at Unilever. Ben returned to school and received his Ph.D. in Chemical Engineering from the University of Colorado Boulder in 2020.
Accelerating the Design of Asymmetric Catalysts with Schrödinger’s Digital Chemistry Platform
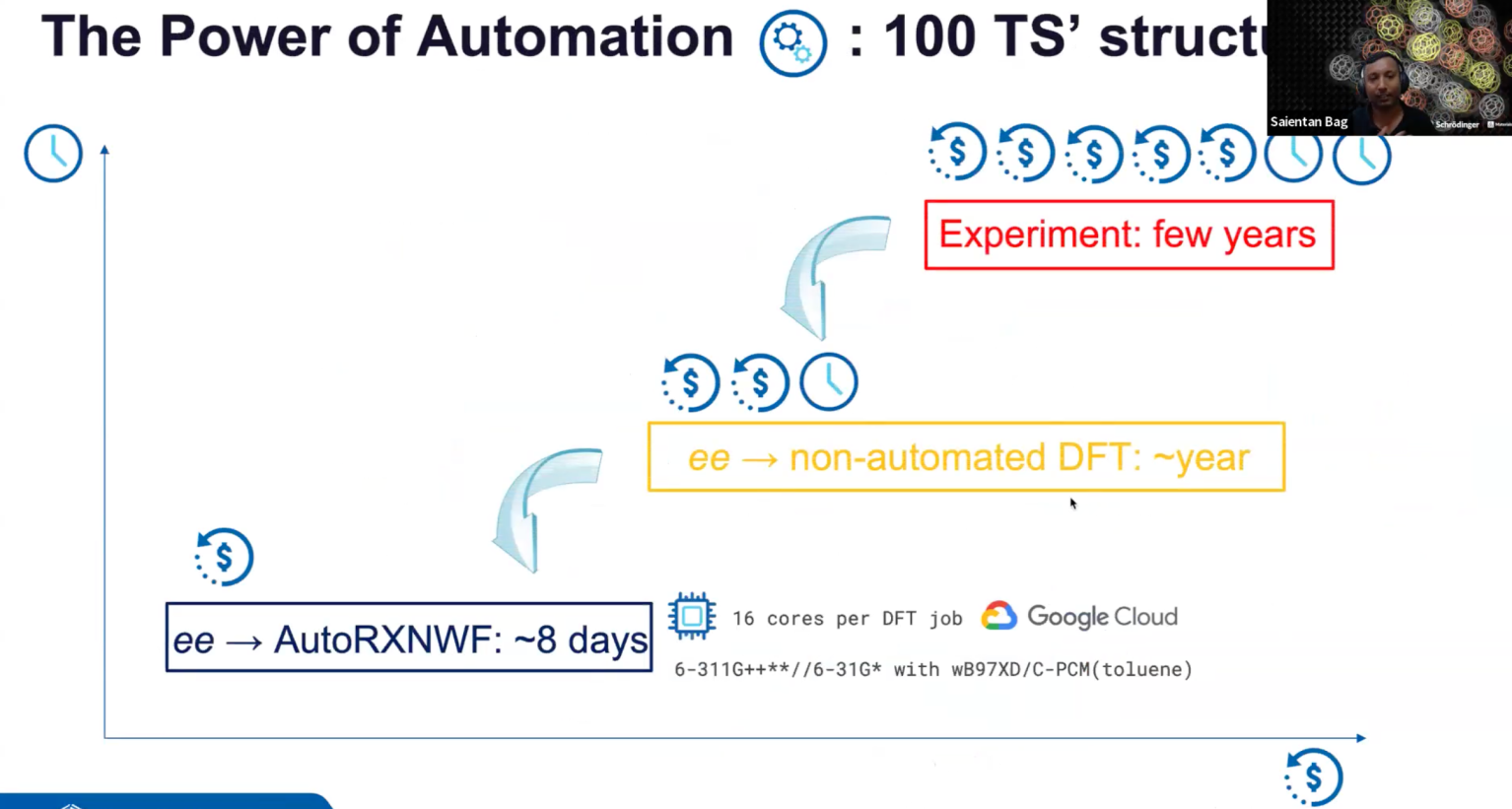
OCT 8, 2024
Accelerating the Design of Asymmetric Catalysts with Schrödinger’s Digital Chemistry Platform
Speaker:
Saientan Bag, Senior Scientist I, Schrödinger
Abstract:
Asymmetric catalysis has become an integral part of the science-driven technological revolution in the second half of the 21st century, leading to decreased energy demands, sustainable chemical processes and the realization of “impossible” transformations. Asymmetric catalysis based on chiral transition-metal complexes plays an important role in the synthesis of single-enantiomer drugs, perfumes and agrochemicals. The importance of the field is recognized by two Nobel Prize Awards in 2001 (transition-metal catalysis) and 2021 (organocatalysis). Asymmetric catalysts are traditionally designed by experimental trial-and-error methods, which are resource-, time- and labor-consuming, and thus extremely expensive. Digital methods offer the opportunity to expedite catalyst design. Until recently, computational chemistry, typically quantum chemical studies, indirectly contributed to asymmetric catalyst design by providing rationalization for the mechanism of generation of chirality. With the development of more advanced methods, algorithms and an included layer of automation, computational catalysis is now providing the possibility for direct asymmetric catalyst design. In this webinar, I will demonstrate how Schrödinger’s advanced digital chemistry platform can be used to accelerate the direct design and discovery of asymmetric catalysts.