Stabilization of myoglobin from different species (produced by cellular agriculture) using food-grade natural and synthetic antioxidants
Nanoscale analysis of plastic contaminants migration in packaging materials and potential leaching into model food systems
Purposeful simulation: Maximising impact in surface chemistry modelling

NOV 12, 2024
Purposeful simulation: Maximising impact in surface chemistry modelling
Join us for a presentation collaboration with Chemistry World, by Simon Elliott, Director of atomic level process simulation at Schrödinger, titled “Purposeful simulation: Maximising impact in surface chemistry modelling.”
Abstract:
The role of materials science in many of today’s technologies hinges on how one material affects another. As a result, a vast body of research is carried out worldwide into surfaces and interfaces. Atomic-scale models of surfaces advance our conceptual understanding, and also deliver quantitative predictions of properties for the design of new materials within devices, formulations and products. With faster hardware and user-friendlier software, these atomic-scale calculations are finding their place in mainstream R&D – both industrial and academic.
In this hour-long, interactive webinar you will learn about a variety of atomistic models of surfaces and gain perspective on the underlying rationale, benefits and limitations of each. Molecular mechanics, quantum mechanics and microkinetics will feature as the main computational engines for interfaces between gases, liquids, solutes, films, electrodes, catalysts and more. This webinar will be suitable for chemistry professionals looking to integrate modelling into their research in a way that delivers tangible scientific results.
Our Speaker

Simon Elliott
Director of Atomic Level Process Simulation, Schrödinger
Characterizing lipid nanoparticle self-assembly and structure using coarse-grained simulations
Characterizing lipid nanoparticle self-assembly and structure using coarse-grained simulations
mRNA/lipid nanoparticle (LNP) technology is the basis for the most successful Covid-19 vaccines and is being adapted for many other types of treatments. A detailed structural understanding of mRNA containing LNPs and their behaviors will aid in implementing and optimizing these therapeutics.
Summary
- Built and validated a coarse-grained model that accurately captures the selfassembly of an mRNA-encapsulating LNP starting from a homogeneous initial condition at low pH and on length-scales comparable with those currently used in therapeutics
- Demonstrated pH-dependent evolution in the distribution of LNP components that is consistent with experiment
- Observed spontaneously formed bleb structures in simulations of RNAcontaining LNPs for the first time

Approach
Schrödinger scientists carried out coarse-grained (CG) simulations using the Schrödinger Materials Science Suite, Desmond molecular dynamics engine, and OPLS4 force field, following the steps below:
- Automated parameterization of CG model for mRNA-containing LNP components using a 1.5 microsecond atomistic reference simulation employing the OPLS4 force field
- Constructed a homogeneous system containing CG mRNA, lipids (with molar composition 45.6/9.4/43.4/1.6 mol % of ALC-0315/DSPC cholesterol/ALC-0159), and water using the new CG model
- Relaxed the system and conducted Langevin dynamics (LD) simulations to investigate evolution of LNP morphologies
- Conducted LD simulations with progressively higher levels of ionizable lipid (ALC-0315) neutralization starting from the final self-assembled LNP, in order to study the changes in internal morphology as the pH rises from the acidic LNP formation conditions to the pH characteristic of blood
Outlook
This work by Grzetic et al. demonstrates that self-assembly of LNPs for nucleic acid delivery can be simulated. The predictive power of the presented model opens the possibility of simulating larger systems with longer RNA sequences and enables investigation of the dependencies of LNP structure on lipid composition and other factors, rapidly providing information that can complement both experimental efforts as well as atomistic simulations.

Characterization of the internal structure of the LNP with 90% of the ALC-0315 protonated at low pH (panels a-c) and with ALC-0315 completely neutralized at neutral pH (panels d-f). Panels b and e show radial density profiles measured from the LNP center, and panels c and f show radial density profiles measured from RNA backbone sites.*
*Reprinted with permission from Mol. Pharmaceutics 2024, 21, 4747-4753. Copyright 2024 American Chemical Society.
Publication
-
Coarse-Grained Simulation of mRNA-Loaded Lipid Nanoparticle Self-Assembly
Grzetic DJ, et al. Mol. Pharmaceutics 2024, 21, 4747-4753.
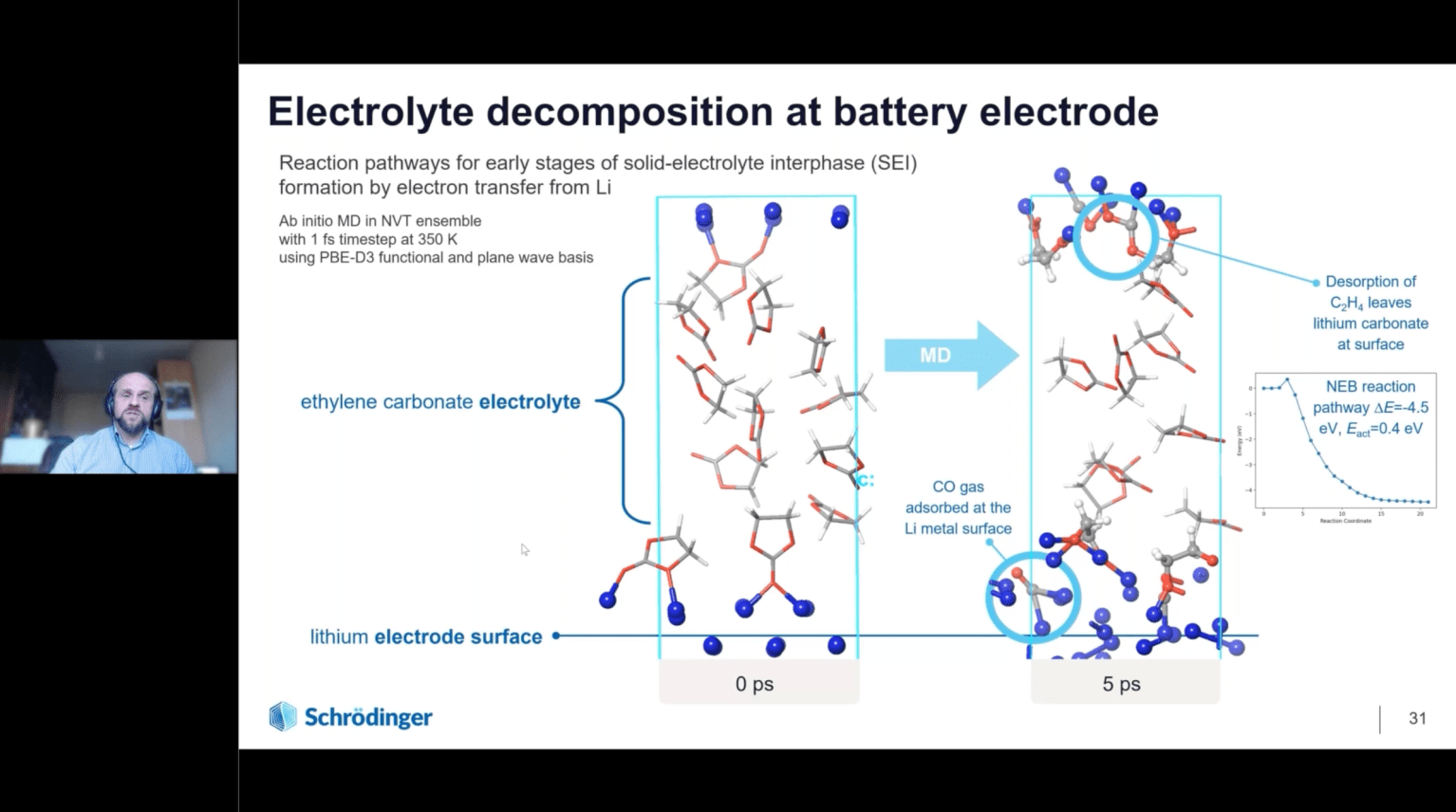
How Physics-based Modeling and Machine Learning Enable Accelerated Development of Battery Materials

OCT 1, 2024
How Physics-based Modeling and Machine Learning Enable Accelerated Development of Battery Materials
Speaker:
Garvit Agarwal, Senior Scientist II, Schrödinger
Abstract:
The rapid advancements in rechargeable Li-ion battery (LIB) technology over the last decade has revolutionized several key industries such as transportation and consumer electronics. However, new battery chemistries are needed to meet the rapidly growing demand and to improve the power density, safety, reliability, and lifetime of LIBs. Molecular modeling has become an integral part of the design cycle of new battery chemistries. Accurate physics-based modeling enables rapid evaluation and screening of large chemical and material design space thereby, helping industries reduce the time required to bring the new technology to the market. In this webinar, we will introduce the latest technological innovations in Schrödinger’s digital chemistry platform for battery materials design. In particular, the webinar will focus on examples to demonstrate the application of automated solutions for accurate prediction of thermodynamic stability and voltage profile of cathode materials, ion diffusion pathways and kinetics in electrode materials, transport properties of liquid electrolytes and modeling the nucleation and growth of solid electrolyte interphase (SEI) layers using Schrödinger’s SEI simulator module. We will also introduce an automated generalized framework for the development of customized machine learning force fields for complex materials such as liquid electrolytes, inorganic cathode coatings and solid polymer electrolytes, paving the way for efficient design of novel materials for next generation batteries.
AI in Drug Discovery USA 2024

Conference
AI in Drug Discovery USA
- October 21st-22nd, 2024
- Boston, Massachusetts
Schrödinger is excited to be participating in the AI in Drug Discovery USA conference taking place on October 21st – 22nd in Boston, Massachusetts. Join us for a presentation by Karl Leswing, Executive Director, Machine Learning at Schrödinger, titled “Latest advancements in machine learning-enhanced in silico design: Impact on a pipeline of drug discovery programs.”
Speaker:
Karl Leswing, Executive Director, Machine Learning, Schrödinger
Key Learning Objectives:
- Using active learning with FEP+ for large-scale in silico fragment screens in hit discovery
- Applying de novo design workflows for intelligent molecular core design
- Leveraging experimental data for enhancing ADMET profiles in lead optimization using an interactive ML dashboard

Karl Leswing
Executive Director, Machine Learning, Schrödinger
Karl Leswing is the Executive Director for Machine Learning at Schrödinger. In this role he oversees the research and execution of machine learning applications for Schrödinger’s digital chemistry platform. In 2017 he was a visiting researcher at the Pande Lab working on using deep learning techniques for drug discovery. During that time he co-authored MoleculeNet, a benchmarking paper analyzing machine learning techniques for chemoinformatics. Karl received his undergraduate degree from the University of Virginia, and a Master’s in machine learning from Georgia Tech.
New York City Integrative Structural Biology Symposium

Conference
New York City Integrative Structural Biology Symposium
- October 9th-11th, 2024
- New York, New York
Schrödinger is excited to be participating in the New York City Integrative Structural Biology Symposium conference taking place on October 9th – 11th in New York, New York. Join us for a workshop and presentation by Schrödinger scientists.
Bring your biochemistry to life with 3D molecular visualization and movie making
Speaker:
Thomas Stewart, Senior Developer, Schrödinger
Abstract:
PyMOL has long been the preferred choice of hundreds of thousands of scientists worldwide for its unparalleled visualization quality, speed, and flexibility.
With PyMOL 3, we’re taking molecular visualization to new heights – empowering scientists, educators, marketers, and communicators to bring their science to life.
Structure-based drug discovery at Schrödinger: At the interface of computational and experimental sciences
Speaker:
Zach Johnson, Director, Schrödinger
Abstract:
Schrödinger’s physics-based computational platform enables highly accurate in silico predictions of key molecular properties, reducing the overall time and cost of drug discovery campaigns by facilitating the rapid exploration of molecules across vast chemical space and decreasing the number of compounds that need to be synthesized and tested experimentally before arriving at a development candidate. The Schrödinger software suite also contains several tools for robust model building and refinement of experimental protein structures. In this talk, we will discuss how experiment and computation come together in our platform to accelerate drug discovery and present examples from our internal programs of our platform in action.
Our Speakers

Thomas Stewart
Senior Developer, Schrödinger

Zach Johnson
Director, Schrödinger
Computational reactivity and catalysis for drug synthesis


SEP 25, 2024
Computational reactivity and catalysis for drug synthesis
Abstract:
Efficient pharmaceutical synthesis is key to a successful and cost-effective drug development strategy. This webinar will take an in-depth look at how computational modeling is transforming pharmaceutical synthesis. We will showcase Schrödinger’s Materials Science platform and its role in studying degradation, reactivity, and catalysis relevant to small molecule active pharmaceutical ingredients. We will examine automated solutions for predicting bond dissociation energies and decomposition products, as well as tools for elucidating reaction mechanisms. The talk will also introduce Schrödinger’s solution for direct, computational homogeneous catalyst design. These powerful yet accessible tools are shaping the future of drug development for both computational and experimental scientists.
Webinar Highlights:
- Introduction to Schrödinger’s Materials Science platform and its application in pharmaceutical synthesis
- Overview of Schrödinger’s quantum mechanical engine (Jaguar) and its basic functionality
- Examples of Nanoreactor and bond dissociation energy calculations for studying active pharmaceutical ingredient (API) degradation
- Examples of AutoTS and AutoReactionWorkflow for reaction mechanism elucidation
Our Speaker

Michael Rauch
Associate Director of Materials Science, Schrödinger
Dr. Michael Rauch is Associate Director of Materials Science at Schrödinger. Michael earned his Ph.D. from Columbia University in synthetic organometallic chemistry as an NSF Graduate Research Fellow before pursuing a postdoctoral role in organic chemistry at the Weizmann Institute of Science as a Zuckerman Postdoctoral Scholar. Michael has made significant research contributions in areas such as green, sustainable chemistry and homogeneous catalysis, with his work being cited more than 1000 times. Michael is particularly interested in transforming the way that synthetic chemists and traditional R&D organizations utilize molecular modeling via practical education.
Efficient Computation of Process Parameters for Controlling the Chemistry of Deposition or Etch
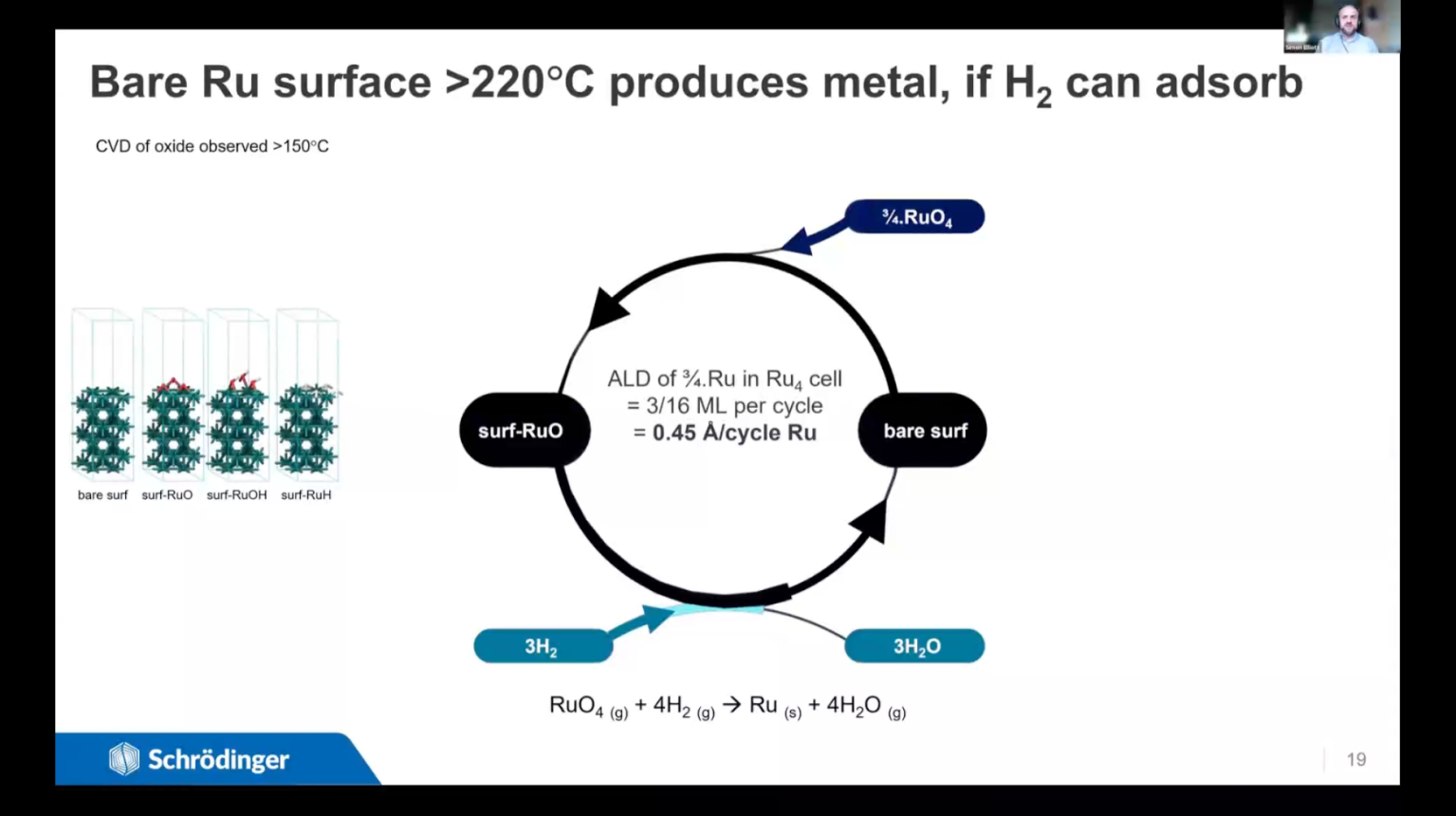
SEP 25, 2024
Efficient Computation of Process Parameters for Controlling the Chemistry of Deposition or Etch
Speaker:
Simon Elliott, Research Leader, Schrödinger
Abstract:
We present a variety of computational techniques for understanding, controlling and improving deposition and etch processes. The emphasis is on choosing the right technique for the research question and time available. The same computational techniques can be used to investigate other gas-surface processes, such as catalysis or sensing. Different chemical processes can be in competition when a solid surface is treated with a gaseous reagent and the outcome is determined by conditions such as temperature and pressure. For instance, continuous deposition (CVD) may take over from self-limiting deposition (ALD) as the temperature is raised. Or temperature may dictate which material is deposited; in the case presented here, ruthenium oxide film is deposited from RuO4+H2 in experiments at 75°C, whereas Ru metal is obtained at 100°C and above. Ru is being investigated as an electroplating seed layer in electronics, as a capacitor electrode and as a heterogeneous catalyst – all applications that require metal rather than oxide. We show that thermodynamics based on density functional theory (DFT) is a computationally-efficient approach for distinguishing between the possible surface-gas processes. The temperatures and pressures for crossover between different chemistries can be estimated, with the accuracy depending on how entropy, coverage and diffusion are treated. We use DFT to examine the conditions of stability for Ru metal, hydride, hydroxide and oxide with respect to H2 and RuO4 reagents, and so explain the crossover from oxide to metal film just below 100°C. We point out how to balance the cost (in terms of researcher time and computer time) against the benefit that each level of accuracy can offer. In the second part of the talk, we introduce Microkinetic Modelling, a new Schrödinger capability for examining the overall kinetics of gas-surface chemistry by solving the coupled kinetic rate equations of its constituent elementary reaction steps. This allows the simulation of macroscopic parameters such as sticking coefficients that can be experimentally measured and used as inputs for fluid dynamics simulations. We first outline the computational scheme, where elementary steps and their activation free energies have been computed with DFT. The resulting microkinetic model for alumina ALD yields measurable quantities (e.g. growth rate) as a function of temperature and pressure, which are validated against experiment. Variation with pressure can account for penetration depth and conformality within high aspect ratio features. The two cases discussed in this talk thus illustrate how atomic-scale DFT can be embedded into higher-level computational schemes for accurate and achievable prediction of the conditions and parameters for controlling chemical processes.
Discovery on Target 2024

Conference
Discovery on Target 2024
- September 30th – October 3rd, 2024
- Boston, Massachusetts
Schrödinger is excited to be participating in the Discovery on Target 2024 conference taking place on September 30th – October 3rd in Boston, Massachusetts. Join us for a poster by Shelby Ellery, Principal Scientist II at Schrödinger, titled “Discovery of first-in-class highly selective Wee2 inhibitors.” Stop by booth #523 to speak with Schrödinger scientists.
Speaker:
Shelby Ellery, Principal Scientist II, Schrödinger
Abstract:
Selective Wee2 kinase inhibitors could address the unmet need for non-hormonal contraception.
In this poster, we describe the identification and further optimization of potent and selective Wee2 inhibitors. Our orthosteric series was improved by using prospective FEP+. In collaboration with HitGen, a DEL screen identified potent allosteric inhibitors. The allosteric series show exquisite kinome selectivity and target engagement in ex vivo mouse oocyte assays.
AAPS 2024 PharmSci 360

Conference
AAPS 2024 PharmSci 360
- October 20th-23rd, 2024
- Salt Lake City, Utah
Schrödinger is excited to be participating in the AAPS 2024 PharmSci 360 conference taking place on October 20th – 23rd in Salt Lake City, Utah. Join us for presentations by Schrödinger scientists. Stop by booth #2503 to speak with us.
Coarse-Grained Modeling of Nucleic Acid-Loaded Lipid Nanoparticle Formulations
Speaker:
Doug Grzetic, Senior Scientist I, Schrödinger
Abstract:
We will start off with a problem statement describing how the complicated nature of lipid nanoparticle formulations makes efficient formulation optimization a challenge. Additionally, the effectiveness of LNP formulations is believed to be strongly correlated to the LNP morphology, but this is difficult to characterize, making predictive, in silico measurements extremely valuable.
Then we will provide a brief description of molecular modeling, emphasizing that for LNP self-assembly length-scales (~100 nm) coarse-grained modeling is required. In addition, high-throughput screening studies require that the building of CG models be automated as much as possible. We briefly review techniques for the automation of this process.
We demonstrate the application of coarse-grained modeling to RNA-encapsulating LNPs, with a case study focusing on the Pfizer-BioNTech COVID-19 vaccine formulation.
Modernize your arsenal of formulation tools with physics-based molecular simulation
Speaker:
Ben Coscia, Principal Scientist I, Schrödinger
Abstract:
The impact of physics-based molecular modeling and simulation on formulation is expanding rapidly with advancement of computer hardware and software algorithms. Cloud-based solutions enable individuals to access the world’s most powerful processors with just an internet connection. Machine learning algorithms continue to be leveraged towards improving the accuracy of our models and to guide high throughput simulation studies towards targeted properties. Despite this progress, one can argue that physics-based simulation is an underutilized technique, in large part due to slow adoption by non-experts. The purpose of this talk is to inform our audience of the accessibility of simulation and empower them to take the first steps towards interrogating their research questions with simulation, with specific emphasis on solubilization. We do this by example, describing two case studies which apply physics-based molecular simulation to gain insight into two different approaches that have direct implications on solubilizing poorly soluble APIs.