A computational-based approach to fabricate Ceritinib co-amorphous system using a novel co-former Rutin for bioavailability enhancement
Transfer learning Lithium and electrolyte potential energy surfaces from pure and hybrid DFT
Machine learning force field ranking of candidate solid electrolyte interphase structures in Li-ion batteries
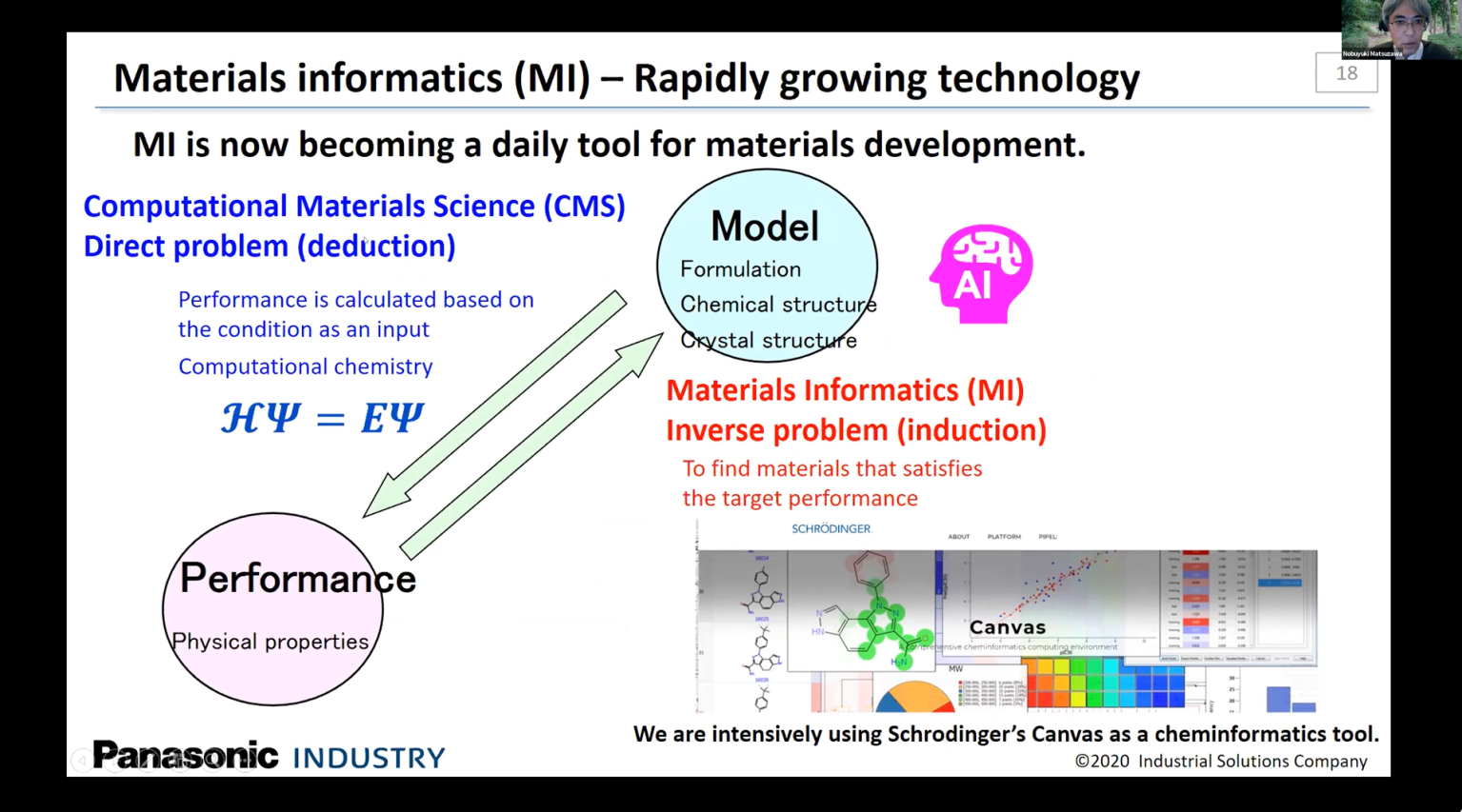
Materials design in electronics industry: Application of materials informatics and cloud computing environment to the design of organic carrier transport materials
OCT 29, 2020
Materials design in electronics industry: Application of materials informatics and cloud computing environment to the design of organic carrier transport materials
Speakers:
Dr. Nobuyuki N. Matsuzawa
Engineering Division, Industrial Solutions Company Panasonic Corporation
Abstract:
In addition to the boosting CPU power owing to the progresses of semiconductor technology, recent expansion of the cloud computing environment is inducing a huge impact on materials design based on computational chemistry by drastically increasing the number of candidate molecules that can be calculated within a reasonable timeframe. Furthermore, rapid progresses in the area of materials informatics (MI) are accelerating the speed of performing prediction of material properties; now a prediction can be made within milliseconds by MI, as compared to hours or even days by the conventional computational methods. These progresses have enabled performing massive screening of millions of materials that might show desired properties. Results of the progresses of recent AI-related technologies are further being introduced to the area of materials development in a form of various proposals to realize the inverse materials design. In this talk, results of our trials to introduce such progresses to the materials design in electronic industry will be presented for the case of the design of organic carrier transport materials such as heteroacenes. Results of quarter million screen of such materials by using the cloud computing environment will be discussed in combination with the results of benchmark studies of various methods of inverse materials design such as deep reinforcement learning.
Molecular modeling of polycyanurates to predict thermophysical properties


OCT 27, 2020
Molecular modeling of polycyanurates to predict thermophysical properties
Speaker:
Dr. Levi Moore
US Air Force, Air Force Research Laboratory Aerospace Systems Directorate, Edwards AFB
Abstract:
Polycyanurates have enjoyed use in high-performance applications in aerospace as the resin component for high-temperature composites. Their high glass transition temperature and low water uptake are two of their most adventitious properties. However, little is known about how water resides in the systems, and despite their low water uptake, failure can occur if a part that has absorbed water experiences a rapid rise in temperature. Modelling and simulation can give insight into the molecular details of water uptake, leading to improved formulations or chemistries that reduce the water uptake of the system, but only if the model realistically represents the experimental system. Two crosslinking mechanisms for realistic construction of a simulated model were evaluated, and the better mechanism was used to prepare systems using varied polycyanurate chemistries. Simulated thermophysical properties like density and coefficient of thermal expansion compared well with literature, while the Tg and water uptake simulations were consistent with literature after correction. Radial distribution function (RDF) analysis showed the absorbed water molecules tended to aggregate in places where there was ample free volume, like around the phenol catalyst, unreacted chain ends, and the triazine crosslink, rather than the bridgehead, where the water molecules were hypothesized to aggregate.
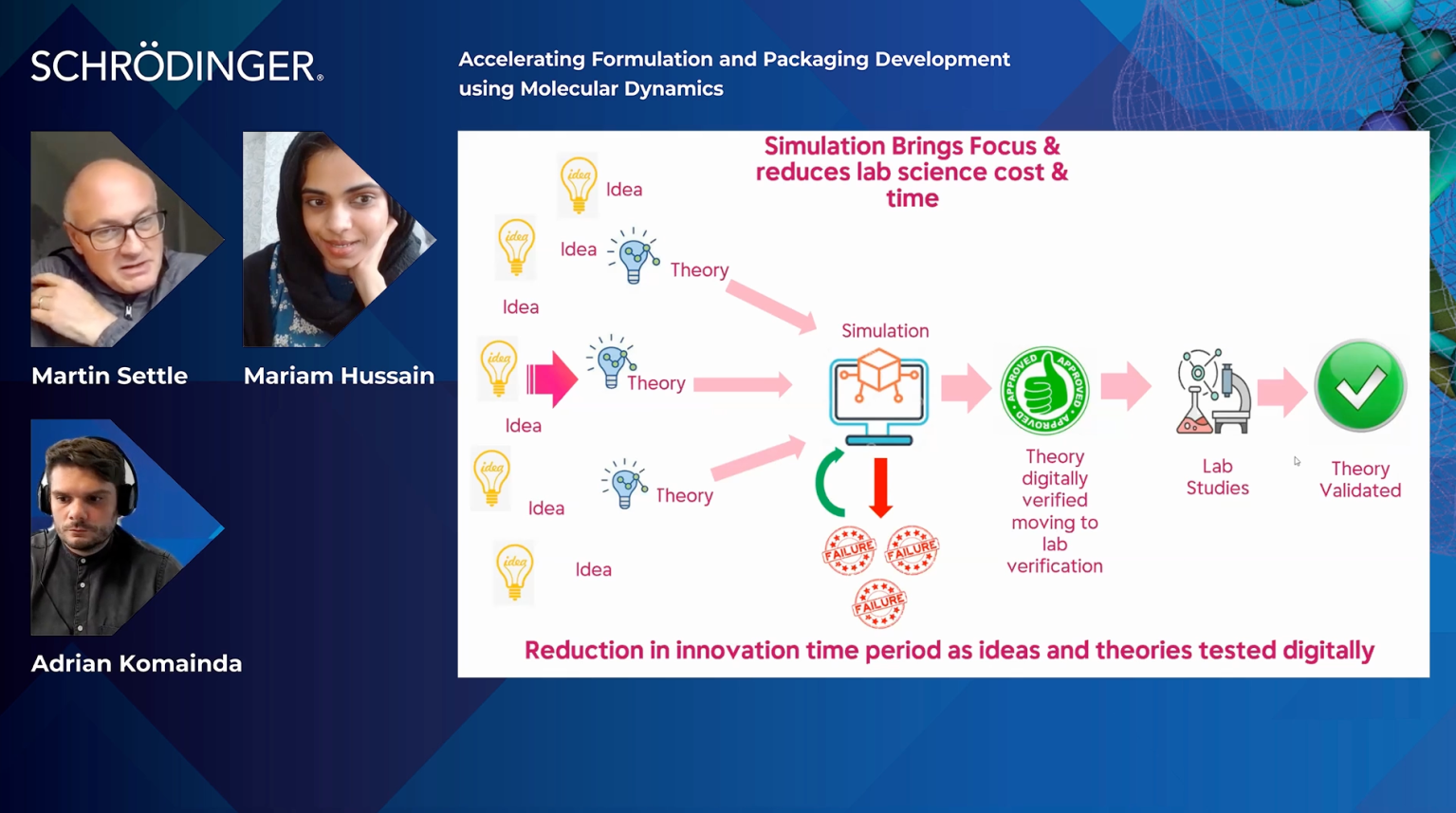
Accelerating formulation and packaging development using molecular dynamics
MAY 10, 2022
Accelerating formulation and packaging development using molecular dynamics
Speakers:
Mariam Hussain, Reckitt Benckiser
Martin Settle, Reckitt Benckiser
Formulation sciences discussion session


MAY 10, 2022
Formulation sciences discussion session
Speakers:
Samuel Kyeremateng, Chris Brown, Afif Faiz-Afzal, John Shelley
Library design: New enumeration tools for lead optimization and idea generation
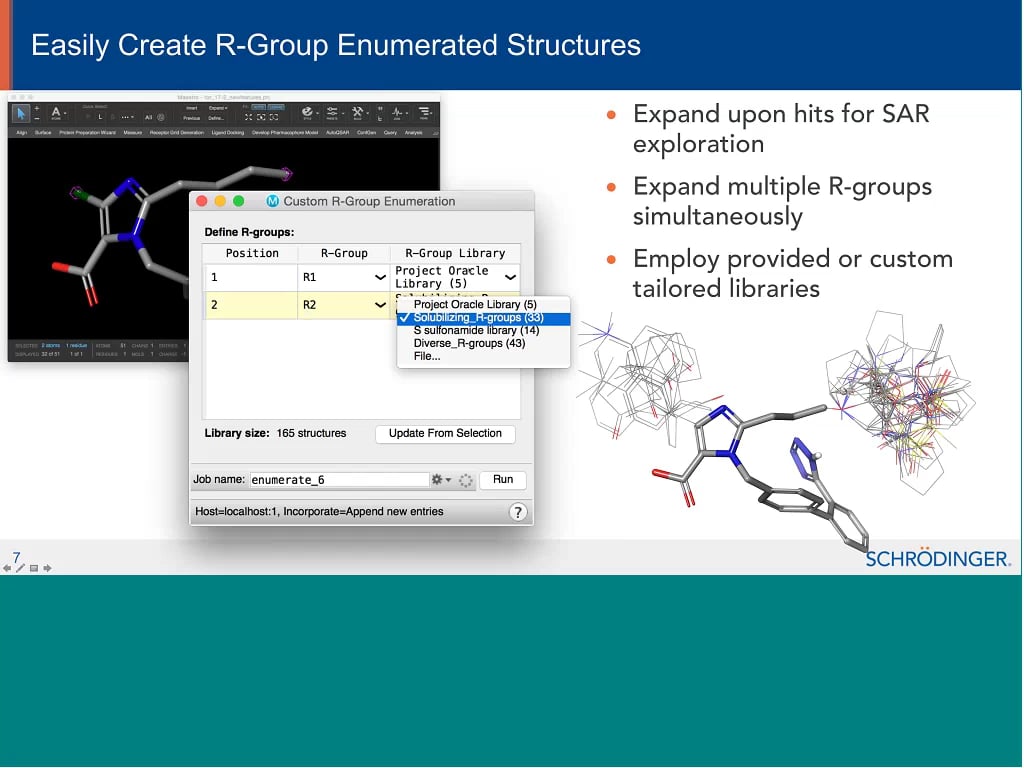
AUG 11, 2021
Library design: New enumeration tools for lead optimization and idea generation
Speaker:
Tatjana Braun
Principal Scientist II
Trends in modern hit discovery: How your ultra-large screens can benefit from machine learning


FEB 2, 2022
Trends in modern hit discovery: How your ultra-large screens can benefit from machine learning
Speaker:
Matt Repasky
Senior Vice President
Abstract:
While traditional structure-based virtual screening has been successful in finding diverse hits to advance projects there is significant room for improvement of hit rates, diversity of hit chemotypes, available IP space explored, and the potency of unoptimized hits. Ultra-large, on-demand synthesizable libraries from vendors have enabled ~100x expansion of purchasable compound space, now billions of compounds, while DNA encoded libraries (DEL) can be even larger. In order to screen these much larger chemical spaces in the billions of compounds, results of two machine learning enabled approaches are described that make it easy and cost effective to find novel hits through virtual and DEL screens of billion compound plus libraries. DNA encoded libraries (DEL) enable screening billions of synthesized compounds but are limited due to high rates of experimental false negatives and positives. Employing machine learning trained to experimental DEL results we demonstrate significantly reduced false negative rates while identifying byproducts in a more favorable property space. To enable efficient, extrapolative chemical space exploration with an accurate docking scoring function, we have developed an active learning-based method employing AutoQSAR/DC machine learning and Glide SP docking as the learner. Results from Active Learning Glide screening of 100 million to billion compound screens show increased chemical diversity and GlideScore of hits relative to brute force screening of subsets of the libraries. Results and costs from these two new methods suggest billion compound library screens could replace smaller, traditional screens commonly employed today.
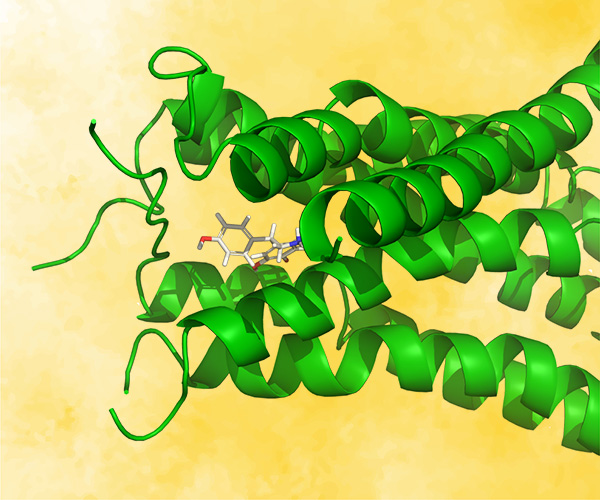
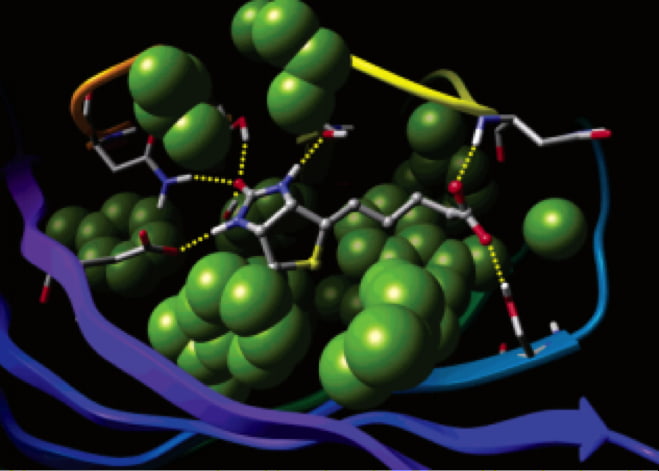
SARS-CoV-2 main protease inhibitors: Physics-based approaches to the discovery of COVID-19 antivirals

JUL 14, 2021
SARS-CoV-2 main protease inhibitors: Physics-based approaches to the discovery of COVID-19 antivirals
Speaker:
Abba Leffler
Senior Principal Scientist
Abstract:
COVID-19 is an infectious disease caused by the SARS-CoV-2 virus. Worldwide, there have been over 240 million cases reported and close to 5 million deaths. Even though there are now several vaccines available for the disease, there is a clear need for the development of small molecule antivirals. One approach to the rapid discovery of such therapeutics is to apply physics-based methods to structures of the SARS-CoV-2 main protease, a key enzyme in the viral replication process. This talk will focus on the medicinal and computational chemistry driven discovery of main protease inhibitors. The challenges of coordinating this multi-company effort, the use of WaterMap to guide the design of inhibitors, and the use of Free-Energy Perturbation (FEP+) to predict the potencies of compounds to prioritize them for synthesis will be discussed. Biological results for key compounds will be presented.
Virtual Screening and Ligand Design using Small-Molecule Drug Discovery Suite

APR 4, 2022
Virtual screening and ligand design using small-molecule drug discovery suite (presented in Turkish)
Speaker:
Dr. Abdulilah Ece