
FEB 11, 2019
Introduction to Schrödinger small-molecule drug discovery suite (presented in Turkish)
Speaker:
Dr. Abdulilah Ece
Moderator:
Rita Podžuna
Senior Principal Scientist
Speaker:
Dr. Abdulilah Ece
Moderator:
Rita Podžuna
Senior Principal Scientist
Speaker:
Agus Rodriguez Granillo
Senior Principal Scientist
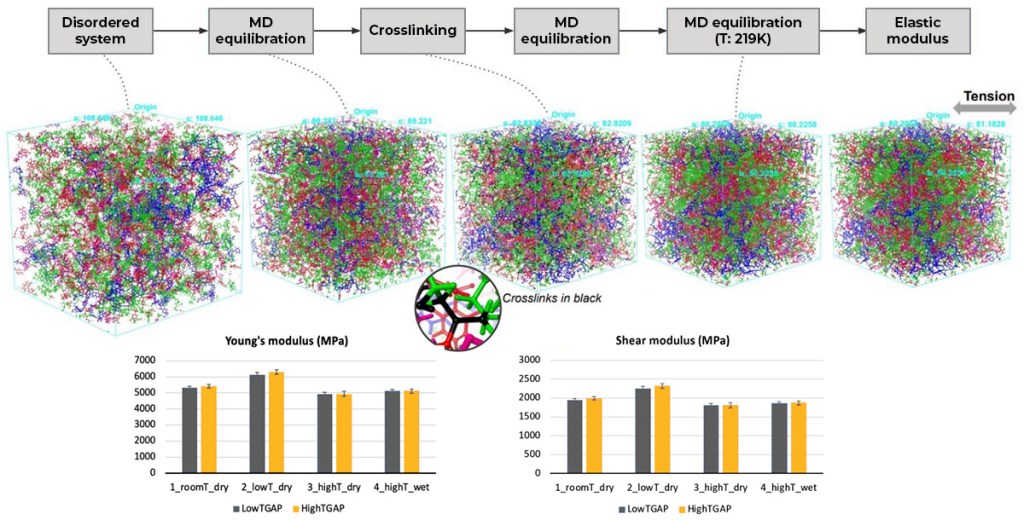
This white paper presents a multiscale modeling framework for designing and characterizing fiberreinforced composites using technologies from Schrödinger and Ansys. By combining molecular modeling, numerical homogenization, and finite element analysis, engineers can predict the effective properties of composites from the length scales of molecular dynamics (MD) to structural analysis. This integrated approach significantly reduces design time and cost while improving the accuracy of material characterization.
The methodology described herein enables virtual screening of chemistries, evaluation of various design parameters, and a deeper understanding of variations in material properties. Benefits include reduced cost, accelerated development, improved design accuracy, and the ability to prototype and optimize products more efficiently. The partnership between Schrödinger and Ansys enables the development and implementation of this multiscale modeling framework, providing engineers with a powerful tool for virtual material identification and characterization.
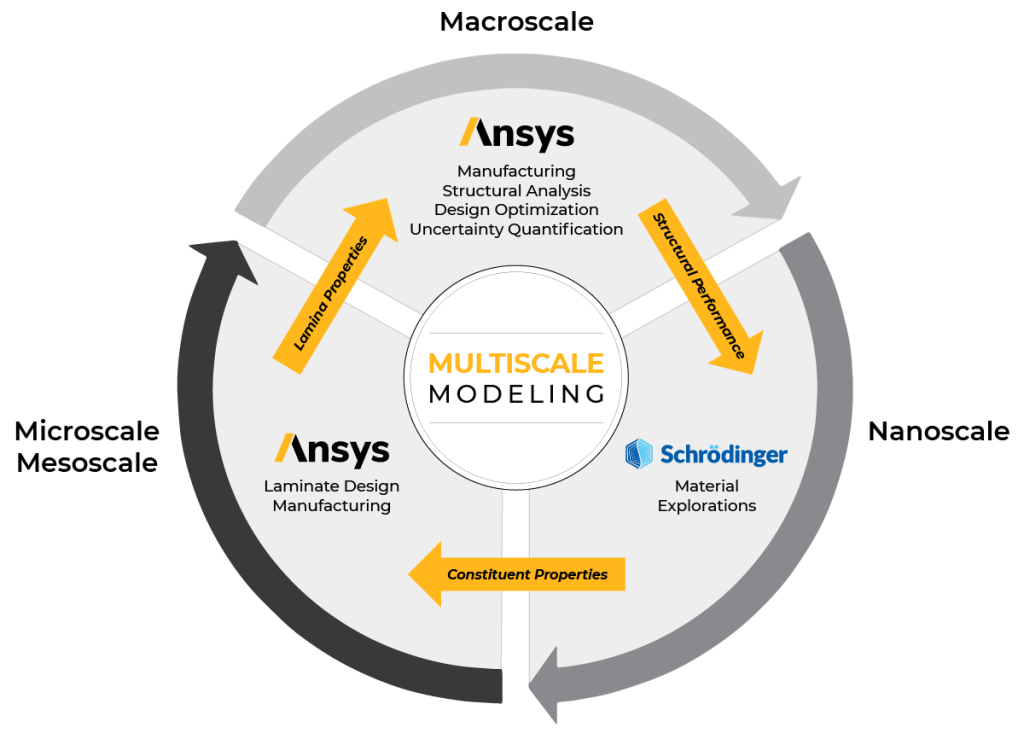

Our world is evolving rapidly and with it comes a wide range of challenges, including the need for sustainable and energy-efficient solutions, advanced electronic devices, and durable, lightweight materials for transportation, aerospace, and construction. Traditional methods for materials discovery or selection are no longer viable for keeping pace with demands.
In this talk, we will introduce a modern approach to materials R&D using a digital chemistry platform for in silico analysis, optimization, and discovery. The platform enables materials design at-scale across a wide range of applications, including organic electronics, catalysis, energy capture and storage, polymeric materials, consumer packaged goods, pharmaceutical formulation and delivery, and thin film processing.
By combining both physics-based modeling approaches (e.g. DFT, molecular dynamics, coarse-graining) and machine learning, researchers can easily incorporate in silico methods into their day-to-day workflows to expedite R&D timelines. Moreover, automated solutions enable scaling from simple molecular property predictions on a local device to high-throughput calculations on the cloud.
We will present real-world case studies that were performed by both experienced modelers as well as novice experimentalists who are new to digital chemistry approaches.
Key Learning Objectives:

Associate Director
Michael Rauch is an Associate Director at Schrödinger specializing in materials science and education. Michael earned his Ph.D. from Columbia University in synthetic organometallic chemistry as an NSF Graduate Research Fellow before pursuing a postdoctoral role in organic chemistry at the Weizmann Institute of Science as a Zuckerman Postdoctoral Scholar. Michael is particularly interested in green, sustainable chemistry and transforming the way that synthetic chemists utilize molecular modeling via practical education.
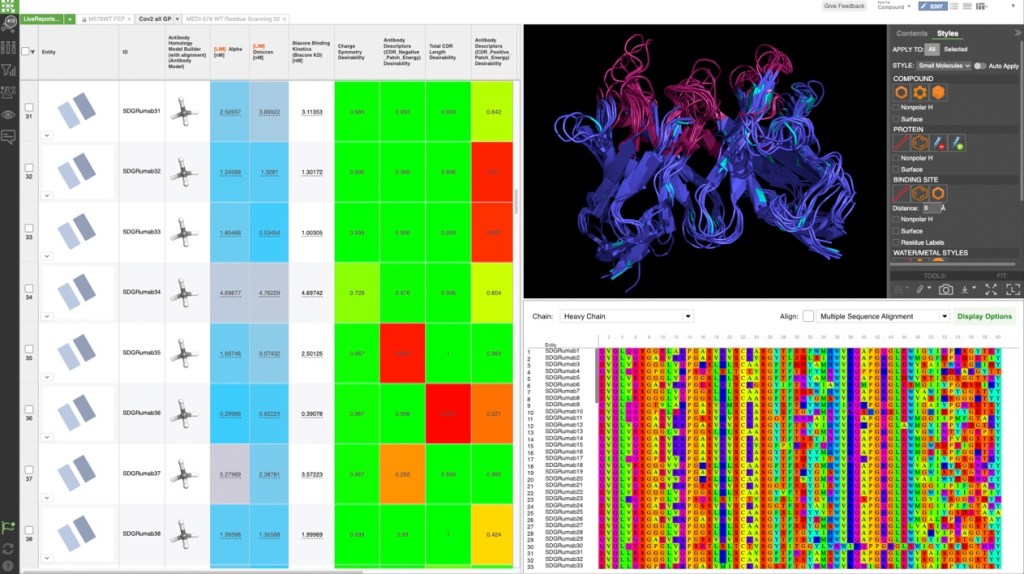
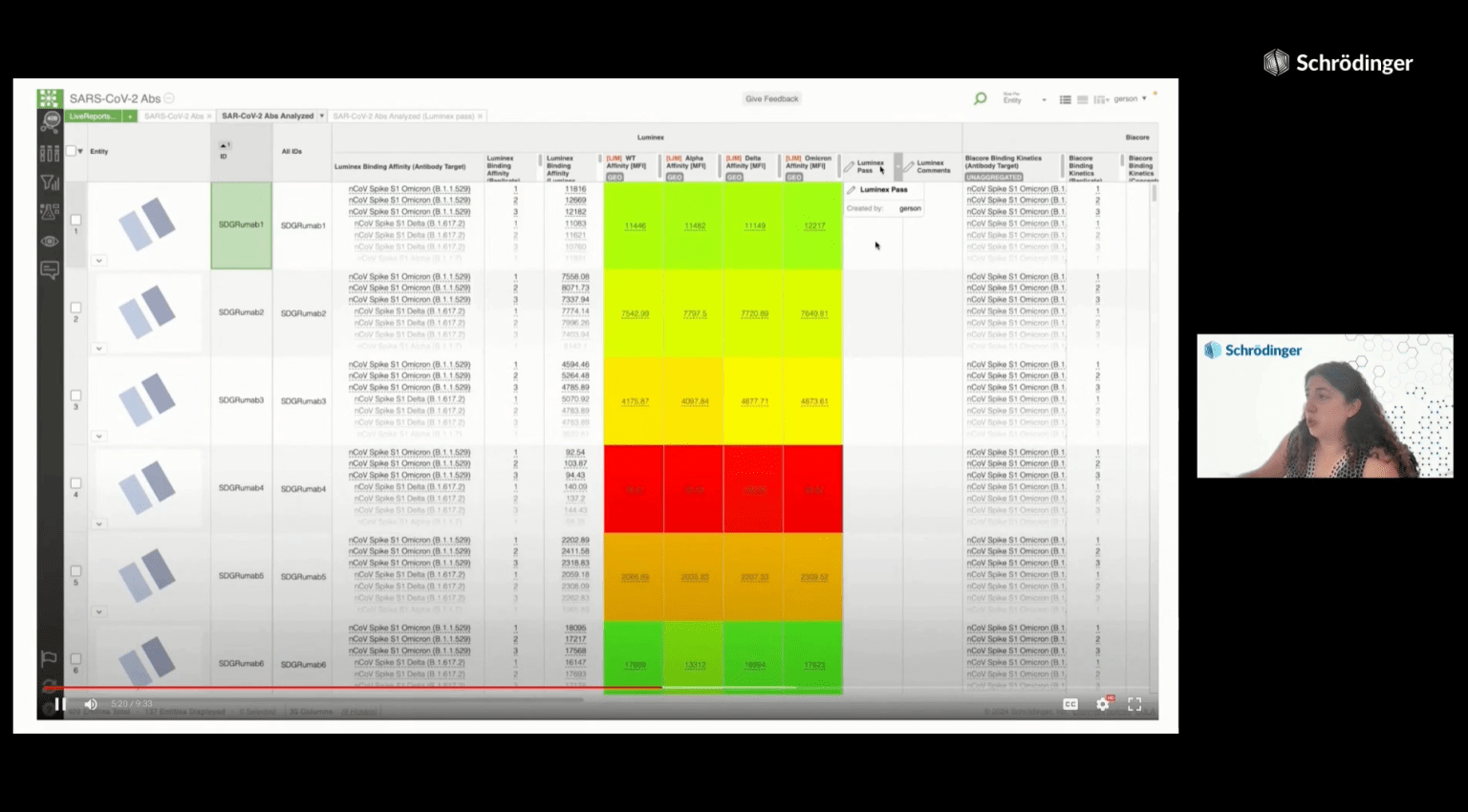
Biologics discovery teams are in need of a comprehensive way to capture and analyze immense amounts of data across all stages of the discovery process. De-siloing this critical information, as well as providing integrated design tools and seamless methods for interdisciplinary communication and collaboration, would allow for more efficient and effective decision-making in therapeutic candidate lead selection and optimization.
This webinar will explore how LiveDesign, Schrödinger’s collaborative enterprise informatics platform, can expedite and improve decision-making in biologics discovery by:
Learn how LiveDesign can accelerate biologics discovery workflows with a software demonstration and live Q&A featuring:

Senior Lead Product Manager, Enterprise Informatics
Cindy Gerson, senior lead product manager, enterprise informatics, joined Schrödinger in 2022. In her role, she leads the LiveDesign for Biologics development efforts – using her extensive knowledge and years of first-hand experience at the lab bench to design software tools that expedite and improve biologics discovery workflows. Prior to joining Schrödinger, Cindy worked at Regeneron Pharmaceuticals in the field of monoclonal antibody-based therapeutics discovery, where she developed, optimized, and executed platforms for the isolation of target-specific antibodies. She completed her BS in Biomedical Engineering at Columbia University and her MS in Bioengineering at Georgia Tech.
半導体や電子部品から日用品に至るまで、あらゆる材料開発において、計算化学は近年では欠かせない基盤技術の一つとしてその存在を確立しつつあります。
シュレーディンガーは、1990年の会社創立以来、分子設計向けソフトウェアとインフォマティクスの機能強化に継続的に取り組み、様々な材料開発の効率化への実用的なソリューションとして提供しています。
この度、これらのソフトウェアを活用した事例をご紹介するセミナーを開催します。
シミュレーションや機械学習のご経験がある方はもちろん、これから手掛けるという方にもご満足いただける内容になっております。
ご興味のあるセッションのみの聴講も大歓迎ですので、ぜひお気軽にご参加ください。
各発表のアブストラクトはこちらからご覧いただけます。
Mohammad Atif Faiz Afzal, Ph.D., Principal Scientist, Schrödinger
シュレーディンガー株式会社 シニア サイエンティスト 青木 祐太(博士(理学))
Haidong Liu, Ph.D., Senior Scientist II, Schrödinger
Pavel A. DUB, Senior Principal Scientist, Schrödinger
Simon D. Elliott, Ph.D., Director – Atomic level process simulation, Schrödinger
Garvit Agarwal, Ph.D., Scientific Lead, Energy Storage, Schrödinger
シュレーディンガー株式会社 シニア サイエンティスト 井本 文裕(博士(工学))
シュレーディンガー株式会社 ストラテジック デプロイメント マネージャー 石崎 貴志(博士(理学))・シニア ソリューション アーキテクト 山田 淳美
シュレーディンガー株式会社 シニア サイエンティスト 大塚 勇起(博士(工学))
【セミナー形式】
Zoom webinarを使⽤したオンライン形式
※海外からの講演者は英語での発表となります。
※Presentations by Japanese speakers are available in Japanese only.
【お申込み⽅法】
▼参加のお申し込みはこちらから▼
https://schrodinger.zoom.us/webinar/register/WN_dbPy5O52RASAIqAp1xfJcQ#/registration
所属企業または所属機関のメールアドレスにて、ご登録をお願いします。
所属が明らかでない、また、個⼈メールアドレスでご登録の場合などは、出席をご遠慮いただく場合がございますのであらかじめご了承ください。参加お⼀⼈様につき⼀登録をお願いします。アクセスリンクの共有はご遠慮ください。
当⽇は、お申し込みの際に登録いただいた⽒名·メールアドレスにてご参加ください。
同業他社さまには参加をご遠慮頂いております。申し訳ございませんが、ご理解のほど宜しくお願い致します。
※ご質問、ご不明な点がございましたら下記までお問い合わせください。
シュレーディンガー株式会社
E-mail: info-japan@schrodinger.com
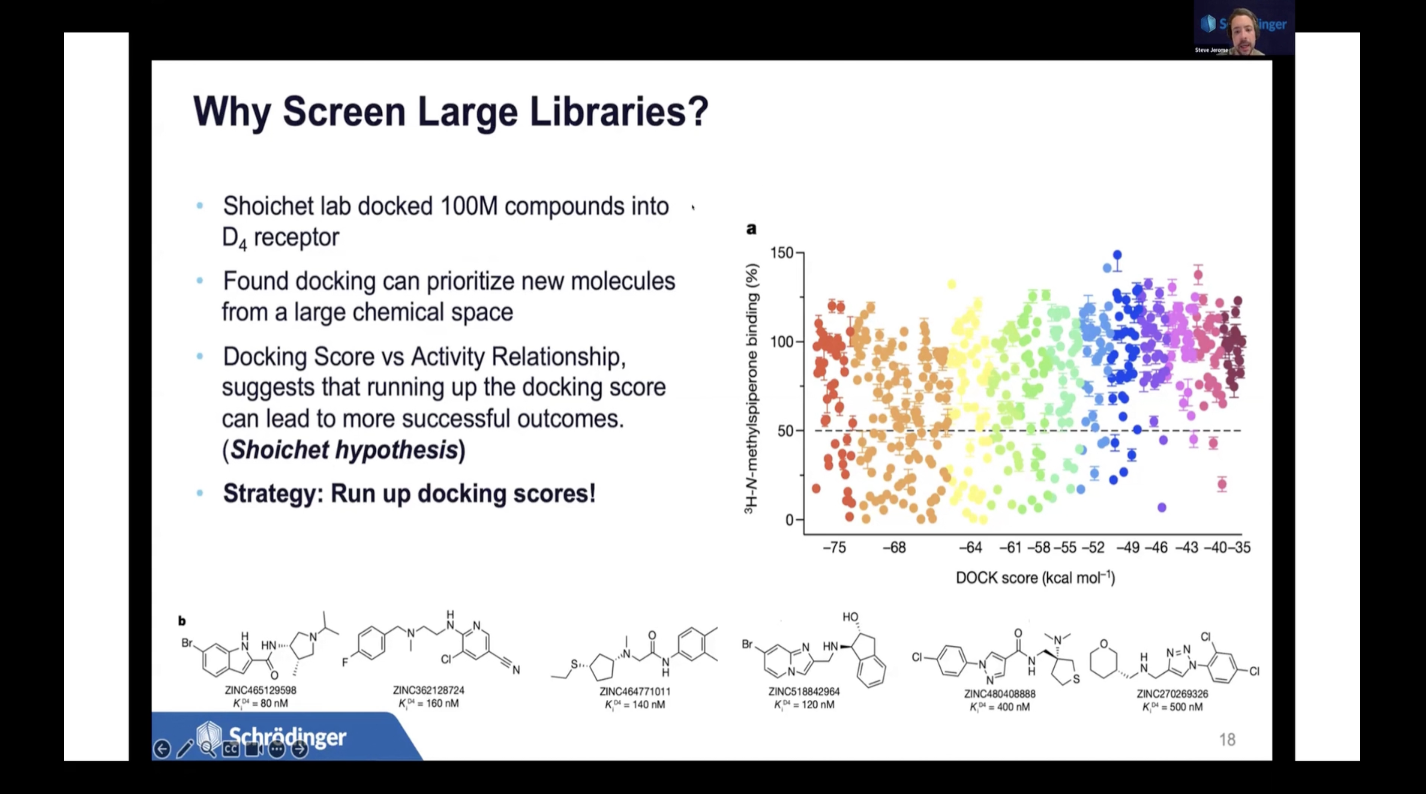
従来のバーチャルスクリーニング手法は、ヒット率の低さや新規性の欠如、そして特定した分子の開発可能性の低さに悩まされてきました。
しかし、シュレーディンガーは、AI/Machine LearningによるアクティブラーニングとAbsolute Binding FEP+を活用した新しいバーチャルスクリーニング手法を開発し、超大規模な化学ライブラリを効果的にスクリーニングすることができるようになりました。性能は大幅に向上し、複数のターゲットにおいて二桁のヒット率を達成しています。
本講演では、この高性能バーチャルスクリーニング手法を用いた、弊社の最新の創薬研究事例を紹介します。

Senior Director
コロンビア大学で化学の博士号を取得。現在はヒットディスカバリー部門のディレクターとして、小分子ヒット同定のための計算ツール開発を指揮しています。
Schrödinger is excited to be participating in the 2024 Workshop on Free Energy Methods in Drug Design conference taking place on May 13th – 15th in Leiden, Netherlands. Join us for a presentation by Robert Abel, Ph.D., Executive Vice President & Chief Computational Scientist at Schrödinger, titled “Expanding the Impact of Free Energy Calculations in Drug Discovery.”

Executive Vice President & Chief Computational Scientist
Robert Abel, Ph.D., executive vice president and chief computational scientist, joined Schrödinger in 2009, is responsible for advancing Schrödinger’s computational science platform. He also leads the computational chemistry team within Schrödinger’s drug discovery group. Robert obtained his Ph.D. from Columbia University, where he was awarded NSF and DHS research fellowships. His thesis work with Professor Richard Friesner involved developing methods to quantify the role of solvent in protein-ligand binding. Robert has co-authored multiple patent applications, and continues to publish extensively on a wide variety of topics in computational chemistry.
Schrödinger is excited to be participating in the 3rd ADDC/ASPET Academic Drug Discovery Colloquium taking place on May 19th – 20th in Arlington, Virginia. Join us for a presentation by Aleksey Gerasyuto, Vice President, Drug Discovery & Head of Chemistry at Schrödinger, titled “The predict-first paradigm: How digital chemistry is shaping the future of drug discovery.”
Speaker:
Aleksey Gerasyuto, Vice President, Drug Discovery & Head of Chemistry at Schrödinger
Date/Time:
May 19 | 4:50 PM
Abstract:
Digital chemistry offers a modern paradigm for enabling rapid in silico testing of design ideas using highly accurate computational assays of key properties, accessible across whole project teams. This shift from design strategies based largely on experimental trial and error towards a ‘predict-first’ approach to drug discovery allows teams to dramatically expand the chemical space that can be explored and results in a highly interactive, computationally-driven design-model-make-test-analyze (DMMTA) cycle. Chemists are empowered to test hypotheses through predictive modeling and iteratively improve designs prior to compound synthesis. Teams can confidently explore novel, and often more complex designs while sending only the top scoring molecules for synthesis. In this talk, we will walk through the digital chemistry strategy used by Schrödinger’s therapeutics group, which has led to successful discovery of clinical candidates SGR-1505 and SGR-3515.
Schrödinger is excited to be participating in the DDF Summit taking place on May 21st – 23rd in Berlin, Germany. Join us for a presentation by John Shelley, Fellow at Schrödinger, titled “Molecular Modeling and Machine Learning for Small Molecule and Biologic Drug Formulation.”

Fellow
John earned a MSc from the University of Waterloo in theoretical chemistry and a PhD from the University of Pennsylvania in computational chemistry. Following post-doctoral research in computational chemistry at the University of British Columbia, he worked for Procter & Gamble studying surfactant structures in solution. For the last 23 years, John has worked for Schrödinger, LLC, as a scientific software developer and a research scientist, managing a number of products including the Materials Science Coarse-Grained product. John has focused on computer modeling of drug formulations for much of the last 8 years.
Selecting and combining the right ingredients in the appropriate manner is essential for successful drug formulation given the inherent challenges and competitive market. With advances in modern machine learning, physics-based simulation techniques and computer hardware, modelling is emerging as a valuable source of information that complements experimental characterization. We showcase a cross-section of capabilities within Schrödinger’s Suite for modeling related to formulations of small-molecule or biologic drugs.
For small-molecule drugs workflows have been created for characterizing crystal polymorphs, crystal morphology and degradation risks as well as calculating elastic constants (bulk modulus, shear modulus, etc.), powder diffraction patterns, glass transition temperatures (Tg), diffusion constants, pKa values, melting points, water adsorption and various solubilities. For biologics our toolset supports homology modeling, and the calculation of aggregation propensity, titration curves, isoelectric points and viscosity among other things.
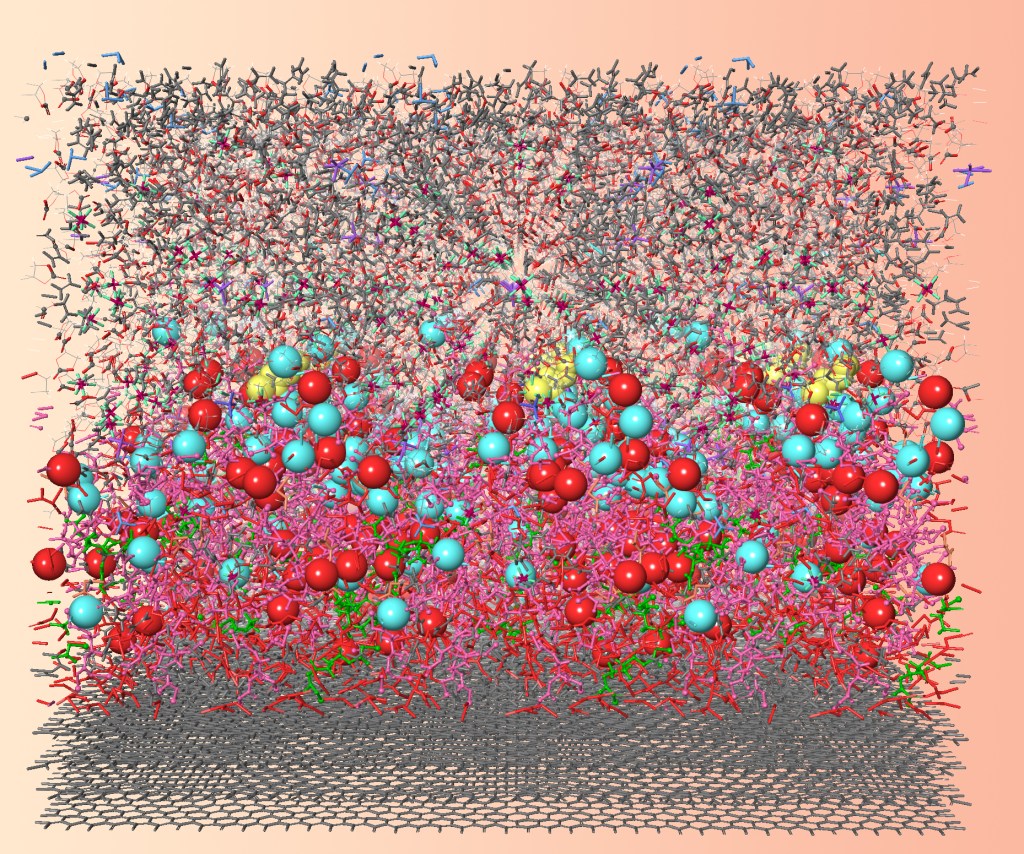
Complex and evolving structures, often in fluid states, play a crucial role in the pharmaceutical industry. For both small-molecule and biologics formulations powerful simulation tools employing atomistic or coarse-grained models to permit the characterization of molecular interactions and nanoscale structuring, sometimes within otherwise disordered bulk systems (e.g., LNP formation, self-assembly of polymer-based structures, dissolving amorphous solid dispersions, liposomes and protein-excipient interactions).
Schrödinger is excited to be participating in the SID Display Week conference taking place on May 12th – 17th in San Jose, California. Stop by booth 1634 to speak with Schrödinger scientists.
Join us for a free workshop day on May 15th in Meeting Room 213. Schrödinger experts will walk you through guided demos and help you gain hands-on experience using digital simulations to expedite your organic electronics R&D. Register for the workshop here.
Speaker: Hadi Abroshan, Principal Scientist, Schrödinger
Speaker: Hadi Abroshan, Principal Scientist, Schrödinger
Abstract: Empowered by digital chemistry and informatics platforms, this study highlights the transformative impact of physics-based simulation, machine learning, and data management in display industry R&D. This technology integration accelerates ideation and decision-making, ensuring swift, accurate, and cost-effective development for next-generation display devices.
Schrödinger is excited to be participating in the Supplier’s Day conference taking place on May 1st – 2nd in New York, New York. Join us for a presentation by Haidong Liu, Senior Scientist at Schrödinger, titled “Molecular modeling of a hair fiber surface by coarse-grained simulation.” Stop by booth 251 to speak with Schrödinger scientists.
Further understanding of the physical properties of the hair surface and its interactions with commonly used ingredients would help to drive new development for hair care products. Molecular simulation can provide an accurate predictive model on the outer layer of the hair to help researchers and engineers understand the fundamental physics at molecular level. In this study, we have built a MARTINI coarse-grained (CG) model focusing on the description of 18-methyl eicosanoic acid (18-MEA). The CG model was derived from an all-atom model but it overcomes the size limits of the all-atom model. We first used the model to characterize the hair surface to understand the distribution of 18-MEA patches. Then the model was used to virtual test the interaction of ingredients on the hair surface. Through modeling of grease molecules and shampoo surfactants on the F-layer of the hair surface, the in-situ cleaning and conditioning process are revealed at molecular scale resolution, which can be correlated to the processes of cleaning and conditioning when washing macroscopically.The MARTINI CG model provides an opportunity to understand the hair surface under different conditions. The unique mechanistic insight of these simulations can help enrich the knowledge of the functioning of the products and help optimize the product performance.