APR 18, 2023
Accelerating the Design of Asymmetric Catalysts with a Digital Chemistry Platform
Speaker
Pavel A. Dub
Senior Principal Scientist
Abstract
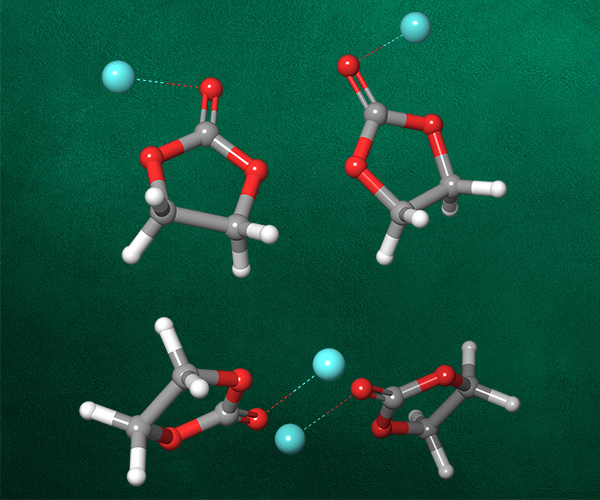
Asymmetric catalysis has become an integral part of the science-driven technological revolution in the second half of the 21st century, leading to decreased energy demands, sustainable chemical processes and the realization of “impossible” transformations. Asymmetric catalysis based on chiral transition-metal complexes plays an important role in the synthesis of single-enantiomer drugs, perfumes and agrochemicals. The importance of the field is recognized by two Nobel Prize Awards in 2001 (transition-metal catalysis) and 2021 (organocatalysis).
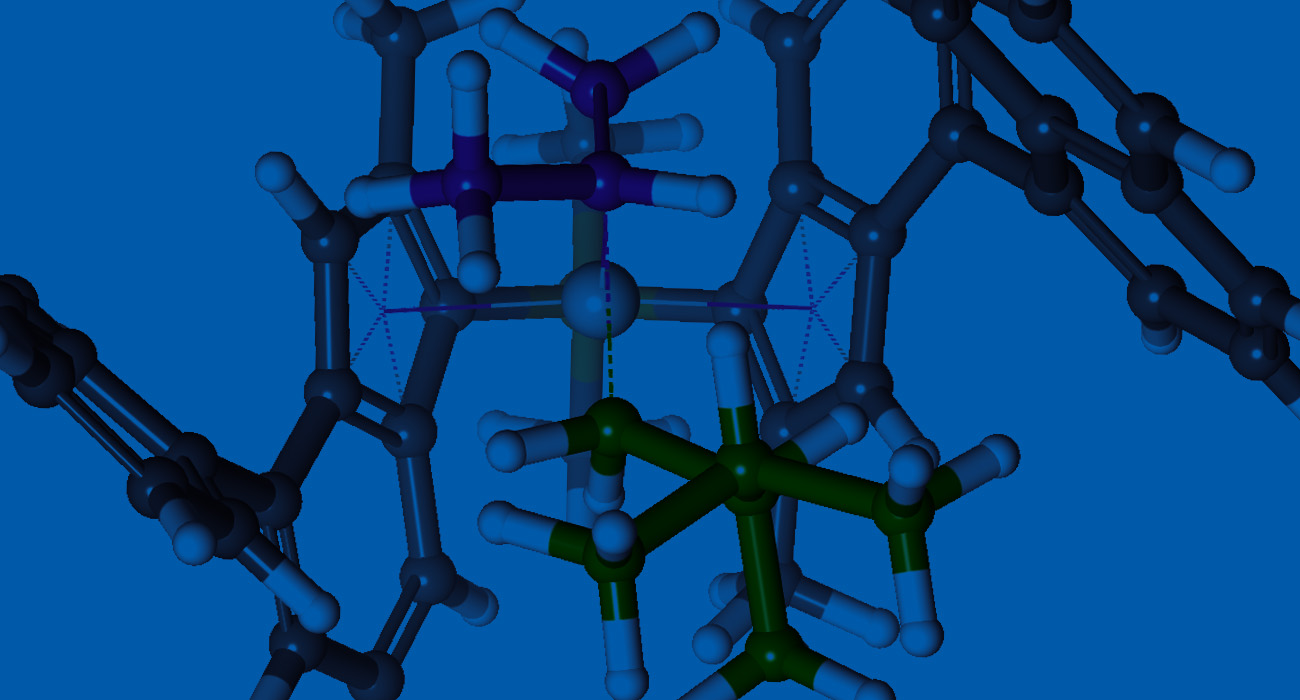
Asymmetric catalysts are traditionally designed by experimental trial-and-error methods, which are resource-, time- and labor-consuming, and thus extremely expensive. Digital methods offer the opportunity to expedite catalyst design. Until recently, computational chemistry, typically quantum chemical studies, indirectly contributed to asymmetric catalyst design by providing rationalization for the mechanism of generation of chirality. With the development of more advanced methods, algorithms and an included layer of automation, computational catalysis is now providing the possibility for direct asymmetric catalyst design.
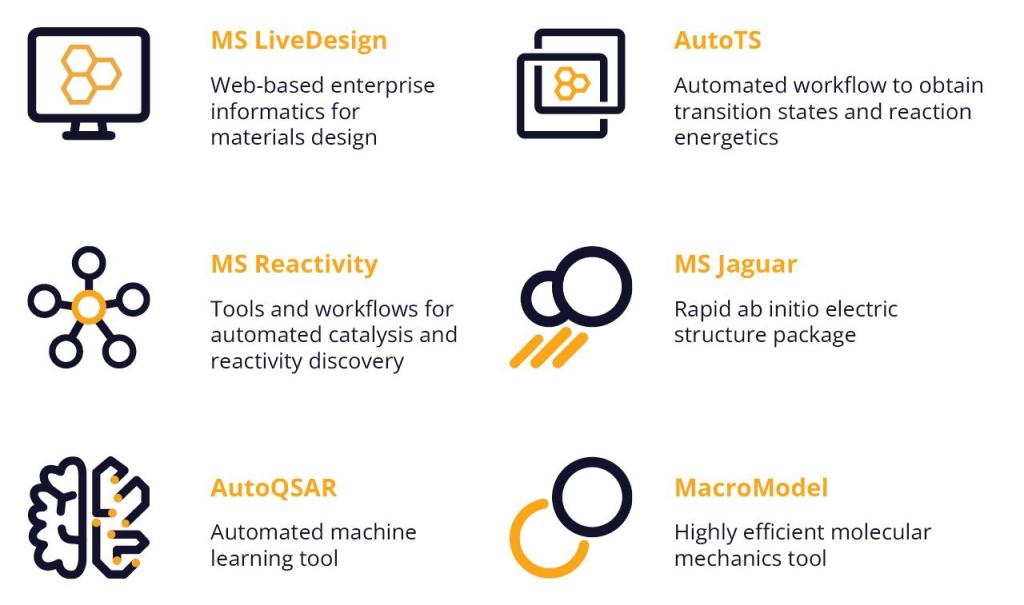
In this webinar, we will demonstrate how Schrödinger’s advanced digital chemistry platform can be used to accelerate the direct design and discovery of asymmetric catalysts.
Key Learning Objectives:
- Learn how to design an asymmetric catalyst with computational chemistry
- Learn how automated high-throughput simulation workflows enable rapid asymmetric catalyst design
- Understand the intersection of physics-based and machine learning techniques in asymmetric catalyst design