Molecular dynamics simulations accelerate the development and optimization of recyclable tire materials
Scientists from Evonik and Schrödinger gain a deeper understanding of the impact of additives and macrocyclic structures on trans-polyoctenamer rubber (TOR).
Accurately predicted key thermophysical and structural properties such as glass transition temperature (Tg), density, free volume, and diffusion coefficient for rubber mixture systems
Explored the miscibility of transpolyoctenamer rubber (TOR) with different crosslinkers and base polymers
Developed an understanding of the impact of macrocyclic polymer chains on the structural and thermophysical properties
Accelerated rational design of highperformance rubber formulations and provided insights into the key factors for processability of recycled rubbers
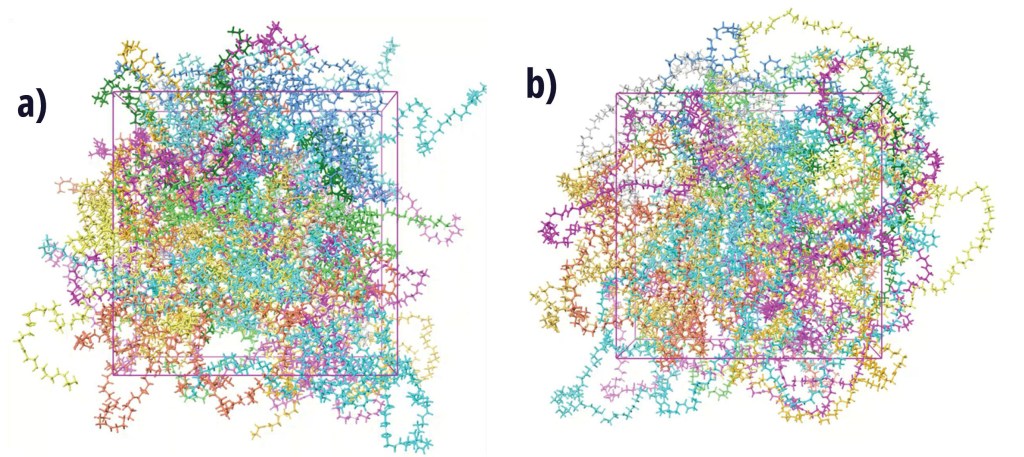
Snapshots of final configuration after the production MD run for a) linear chains, and b) macrocyclic chains of 2000 molecular weight.1
Challenges
Trans-polyoctenamer rubber (TOR), commercially known as VESTENAMER®, has been widely used as a tire additive to improve the processability and recyclability of tire rubbers. In order to efficiently co-vulcanize different rubbers to obtain improved properties, it is vital to understand the interaction of TOR with different components of rubber formulations, including crosslinkers and base polymers. However, experimental characterization of these formulations is time-consuming and resource-intensive.
Approach
Molecular modeling provides an economical alternative to long experiments and lengthy development cycles for new tire formulations. Scientists from Evonik and Schrödinger worked together to digitally evaluate the miscibility of TOR with various crosslinkers and polymers, and to look into the impact of macrocyclic polymer chains on TOR properties. With GPU-accelerated molecular dynamics (MD) engine, scientists were able to run the simulations at high-speed, e.g. 50ns of diffusion calculations, in only a few hours. All models and simulations were performed using the Schrödinger Materials Science Suite and Desmond for MD according to the following procedure:
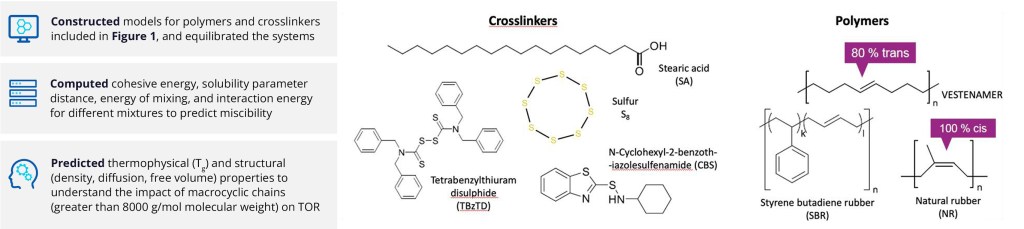
Figure 1. Structures of crosslinkers and polymers used in simulations.1
Results
The computational approach revealed new insights into the compatibility of various polymers/crosslinkers and provided an accurate evaluation of the structural and thermophysical properties of TOR systems as summarized to the right. The work serves as an example of how digital molecular simulations can provide unique insights that are not readily available or economical to study in an experimental setting.
The simulation methods and workflows presented in this work can be easily extended to study other polymer/crosslinker systems.
Summary
The miscibility order for the crosslinkers in SBR, NR, and TOR is TBzTD > SA > CBS > S8, suggesting S8 likely has a limited miscibility in all three polymers, which is also demonstrated by computing the energy of mixing
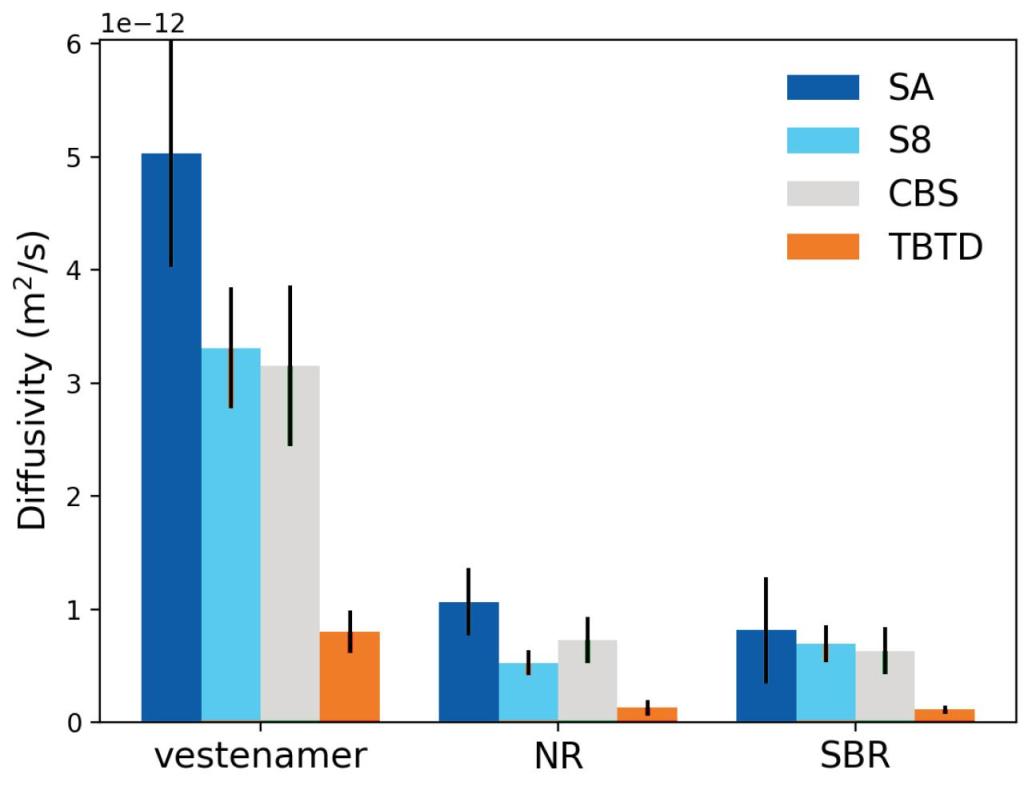
Crosslinkers diffuse more in TOR when compared to SBR and NR allowing for better crosslinking
SBR, NR, and TOR are likely not miscible with each other. A mixture of these three polymers would most likely result in a three-phase system, which is in line with experimental studies
Interaction energy and density analysis indicated ideal mixing at low crosslinker concentration for all the mixtures
TOR macrocycles can potentially affect the viscosity as well as other mechanical properties, and may contribute to lower glass transition temperature of TOR
Figure 2. Predicted diffusivity of different crosslinkers in TOR, NR, and SBR
References
Using molecular simulation to assess the impact of additives and macrocyclic structures on trans-polyoctenamer rubber (TOR) Mohammad Atif Faiz Afzal, Benjamin J Coscia, Andrea R Browning, Thomas S. Asche, Alexander Paasche, Mathew D. Halls International Elastomers Conference, Oct 4-7, 2021, Pittsburgh
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
The pKa of a drug is a key physicochemical property to consider in the drug discovery process given its importance in determining the ionization state of a molecule at physiological pH. Schrödinger provides several solutions for predicting pKa values, protonation state distribution and derived properties that can be applied across a range of drug discovery stages, from screening through lead optimization. Here we provide an overview of each technology solution and use case examples of how they can be applied in drug discovery.
Background
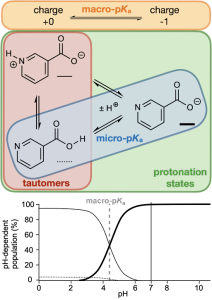
Small molecules can undergo ionization in solution where they either lose or gain protons (H+) at different ionizing sites. The measure of the propensity of a site or molecule to ionize by the association/dissociation of one or more protons is quantified by a pKa value. If the pKa value refers to a particular site ionizable site the value is a microscopic pKa (micro-pKa), and it is a macroscopic pKa (macro-pKa) if the value refers to the entire molecule. The specific arrangement of protons around the ionizing sites constitutes a protonation state, and different protonation states of the same charge level are called tautomers. Each protonation state is in thermodynamic equilibrium with the others and therefore has a free energy associated with its population within this collection of protonation states, which may be derived either from micro-pKa values through thermodynamic equations or obtained directly by comparing the free energies of the states. In drug design, understanding the different protonation states of a molecule is critical, since they will drive properties including solubility, membrane permeability, and activity.
Challenges of pKa Prediction
Determining which states predominate at a given pH and by how much is a challenging task both experimentally and computationally because the number of states that are all in thermodynamic equilibrium grows ~2n with the number, n, of singly protonatable sites. Thus, molecules with many titratable sites can potentially have a large number of different protonation states, all of which need to be enumerated and energetically scored.
Computationally, Schrödinger uses two main approaches to score states: 1) through evaluating thermodynamic equilibrium equations with micro-pKa values, and 2) directly predicting the states’ relative free energies. Predicting pKa values is an important step to calculating state distributions, which in turn enables prediction of important related quantities that would otherwise be inaccessible.
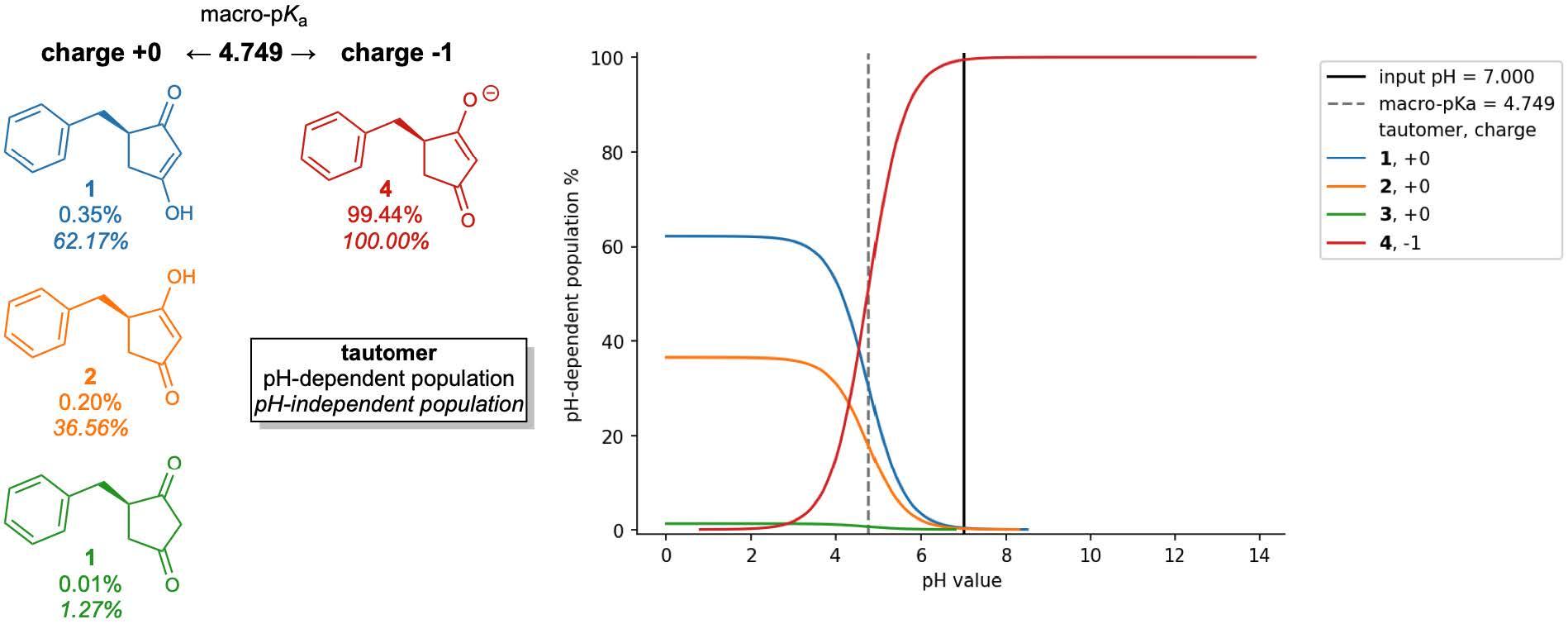
Figure 1: Relationships between macro-pKa, micro-pKa, protonation states, and tautomers and the corresponding speciation diagram.
Overview of Schrödinger Solutions
Epik Classic
Epik Classic, previously known simply as Epik1, is an expert system for rapidly and accurately predicting the micro-pKa values and the most populated protonation states for a ligand at a given pH. The underlying pKa prediction technology is the empirical Hammett-Taft linear free energy relationship (LFER), which identifies an ionizing group, takes its root pKa value, perturbs it by the bonded chemical fragments, and applies charge spreading to arrive at its effective micro-pKa value. Epik Classic then uses the predicted pKa values to enumerate a ligand’s protonation states, rank them by energy, and then return the most populated states. Because Epik Classic uses SMARTS patterns-based rules, it is fast enough for high-throughput, although at the expense of being unaware of both conformational and stereochemical effects.
Epik 7
Epik 7 is a complete redesign of Epik that leverages Schrödinger’s powerful machine learning (ML) technology for more accurate results across broader chemical space. Ionizing groups are initially identified by SMARTS patterns and are then used to enumerate the protonation states for a range of ionizations.2 The micro-pKa values of each site in each state are predicted with 3-layer atomic graph convolutional neural networks (GCNNs) extending out radially six bonds from the ionizing atom. The predicted pKa values for the states are then used to predict the relative energies of the states to both allow determination of the most populated states at a pH and calculation of macro-pKa values. The topological nature of the ML approach means that Epik 7, like previous versions, is rapid but agnostic to 3D geometry and stereochemistry.
Jaguar pKa
Jaguar pKa takes a third, more physics-based approach to predicting micro-pKa values for a ligand. This workflow calculates the pKa values at the user-defined ionizing sites in a query ligand by first generating the conjugate pair, on which are then executed conformational searches to locate the lowest energy structures,3 followed by density functional theory (DFT) based geometry optimizations and single-point energy evaluations. These resulting conformationally-averaged, “raw” micro-pKa values are then corrected using empirically-parametrized relationships to give accurate predictions. Jaguar pKa performs best on non-tautomerizable structures. Being physics-based, it does take into account geometric and stereochemical effects, but at the expense of speed.
Macro-pKa
Macro-pKa follows the same philosophy as Jaguar pKa by combining physics-based DFT calculations with empirical corrections, but extends its applicability to enable calculation of tautomerizable ligands. Macro-pKa automatically identifies ionizing sites, enumerates the protonation states, and calculates the micro-pKa values following a similar workflow to Jaguar pKa, but with an enhanced scheme for generating empirical corrections. Finally, the calculated micro-pKa values are used to rank the protonation states by energy, return the most populated states for a user-supplied pH, and determine the macro-pKa values for the ligand. The exhaustiveness of this approach comes at a larger time and resource cost than Jaguar pKa.
Use Cases
Here we outline several use cases for pKa prediction in the drug discovery workflow.
Note: Each use case example outlines below could be approached with any of the listed solutions within that section. The dataset presented highlights the applicability of just one of the possible solutions.
I. Querying microscopic pKa values
Applicable Solutions
Epik Classic
Epik 7
Jaguar pKa
When investigating the binding modes of a ligand, the micro-pKa value of an ionizing site is an indicator of the propensity for it to become ionized at a given pH. The ionization state of the ligand directly influences how it interacts with another molecule such as a protein, e.g., whether or not it can participate in a salt bridge.
Figure 2: Jaguar pKa micro-pKa predictions for a dataset of small molecules.
II. Querying apparent or macroscopic pKa values
Applicable Solutions
Epik 7
Macro-pKa
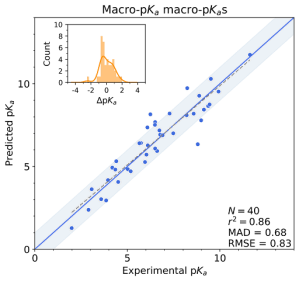
For monoprotic or polyprotic compounds with a single dominant tautomer at each charge level, micro-pKas may very closely match the apparent or macro-pKa value that is most commonly obtained through titration experiments. However, for compounds or ionization states with multiple competitive tautomers, the micro-pKa value of a single tautomer may not fully reproduce the experimentally observed macroscopic value. To obtain this apparent value, all states’ must first be enumerated and evaluated so that all their micro-pKa values are considered in the macro-pKa calculation.
Figure 3: Macro-pKa macro-pKa predictions for a dataset of tautomeric molecules.
III. Ligand preparation and high-throughput screening
Applicable Solutions
Epik Classic
Epik 7
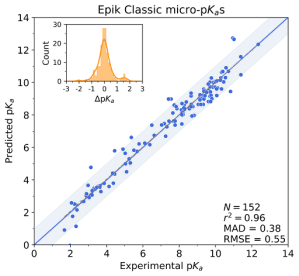
Physics-based simulations typically require specification of all atoms in the simulation system, including all hydrogen atoms. Thus, structure-based simulations including Glide docking, molecular dynamics, and free energy perturbation with FEP+ should be performed using an ensemble of the highly-populated protonation states of a ligand. Therefore, a crucial first step in any structure-based screen of a small molecule ligand library is to prepare the ligands by obtaining the most populated protonated states. Epik Classic and Epik 7 are integrated with our automated ligand preparation workflow, LigPrep, to allow preparation of large ligand libraries for high-throughput screening. Additionally, both Epik Classic and Epik 7 and their LigPrep implementations allow for the generation and scoring of additional states that may potentially bind to metal ions in the pocket.
Figure 4: Epik Classic micro-pKa predictions for a dataset of 152 drug molecules
IV. Hit-to-lead optimization
Applicable Solutions
Epik Classic
Epik 7
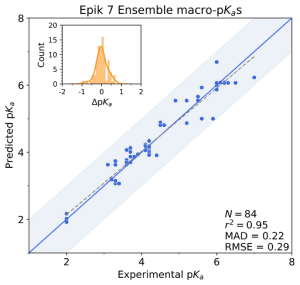
Once hits are identified, a series of analogs are synthesized to explore the relevant chemical space in greater detail to arrive at improved behavior. It is important to be able to screen potential candidates rapidly and accurately to assess which to optimize further. The < 0.5 log unit accuracy and sub-second calculation speed of Epik Classic and Epik 7 make them excellent tools for rapid idea generation and testing. In addition to pKa value and protonation state distribution prediction, they have been implemented in other ADMET or property predictors, such as for membrane permeability and solvation energy.
Figure 5 Epik 7 macro-pKa predictions for a dataset of congeneric tricyclic thrombin inhibitors.
V. Early-stage lead optimization
Applicable Solutions
Epik 7
Jaguar pKa
Macro-pKa
Optimizing the many physical characteristics required can be laborious and costly, from ideation, through synthesis and assay. In this environment, where high quality property predictions are required and time permits, Schrödinger’s physics-based predictors, Jaguar pKa and Macro-pKa, take into account more molecular characteristics, including conformational and stereochemical effects to improve pKa prediction accuracy. Additionally, Macro-pKa and Epik 7 both offer detailed speciation reports for a queried ligand. These are especially helpful for understanding the distribution of tautomeric states across the pH spectrum.
Figure 6: A Macro-pKa report detailing the macro-pKa value and the distribution of protonation states across a pH range.
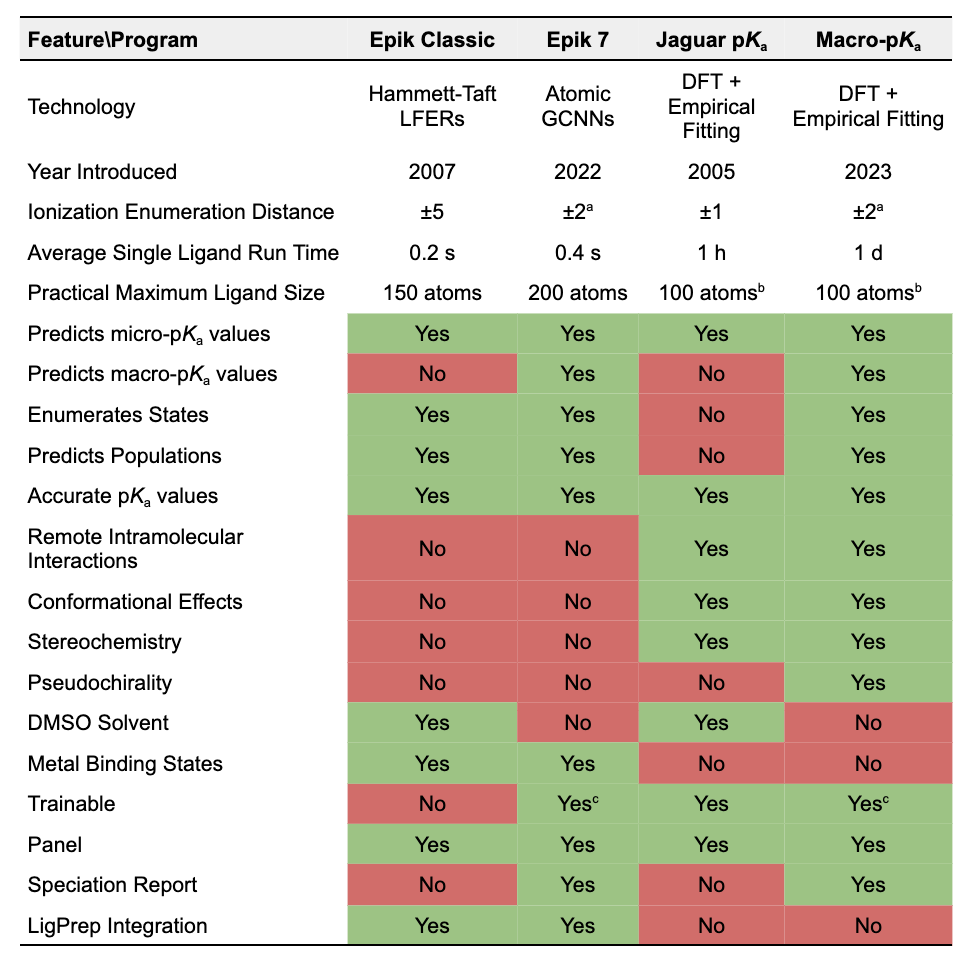
Feature Comparison Table
a) Easily adjustable; b) Strongly influenced by the number of conformers (and tautomers in Macro-pKa); c) Only by internal experts at this time. Table 1. Comparison of Features of the Small Molecule Protonation State Enumeration and pKa Prediction Technologies
References
Epik: A Software Program for pKa Prediction and Protonation State Generation for Drug-like Molecules.
Shelley, J. C. et al. J. Comput. Aided Mol. Des. 2007, 21 (12), 681–691
Epik: pKa and Protonation State Prediction through Machine Learning.
J. Chem. Theory Comput. 2023, 19 (8), 2380–2388
Multiconformation, Density Functional Theory-Based pKa Prediction in Application to Large, Flexible Organic Molecules with Diverse Functional Groups.
Bochevarov, A. D. J. Chem. Theory Comput. 2016, 12 (12), 6001–6019.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
“By combining Morphic’s integrin drug hunters with Schrödinger’s in silico molecular design technologies, we were able to successfully deliver a novel small molecule with the key potency, selectivity, and PK properties for this highly validated biological target where others had previously failed.”
Matt Bursavich VP, Head of Chemical Sciences
Morphic Therapeutic
Design challenge
α4β7 integrin is a well validated target for the treatment of inflammatory bowel diseases (IBD), such as Crohn’s disease and ulcerative colitis. The approved biologic vedolizumab is an antibody targeting α4β7 and is delivered via injection or infusion, severely limiting accessibility and affordability. An orally bioavailable small molecule therapeutic would represent an enormous step forward in addressing these diseases on a global scale. Despite R&D efforts for over 20 years, the design of potent and selective small molecule inhibitors of α4β7 has been unsuccessful due in large part to the daunting task of overcoming the protein’s structural similarity to other integrin family members. Furthermore, maintaining high potency while balancing optimal physical properties in the design of an oral small molecule inhibiting protein-protein interactions is a formidable challenge.
Morphic Therapeutic, experts in all things integrin, partnered with Schrödinger’s drug discovery team to design an orally bioavailable drug selectively targeting α4β7 integrin to treat inflammatory bowel disease. The goal of the program was to leverage a digital chemistry approach, using the power of physics-based predictive modeling technologies combined with Morphic’s unique integrin biology and chemistry insights, to design a highly selective α4β7 inhibitor with best-in-class properties.
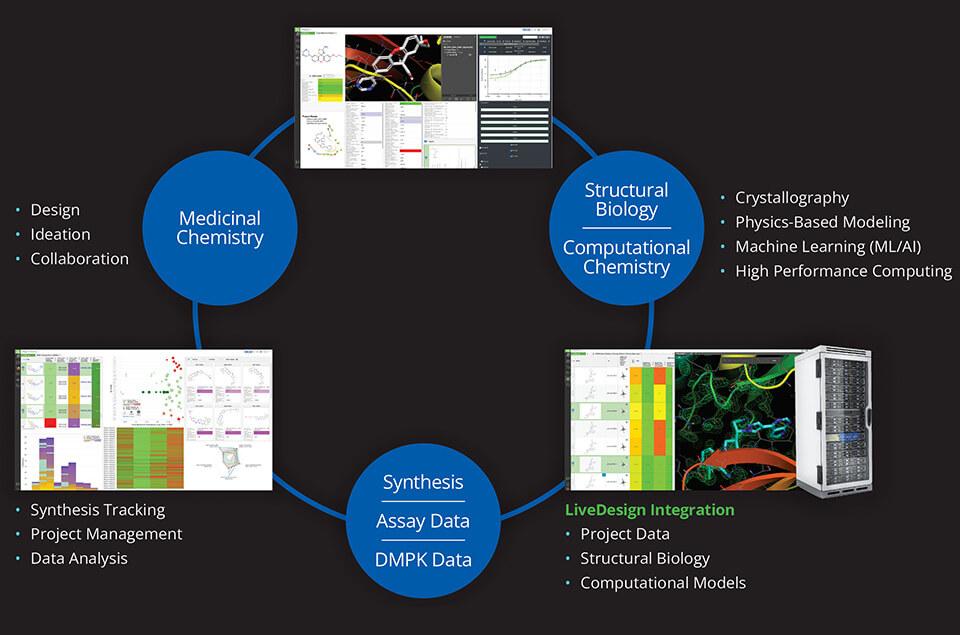
Driving efficient, data-driven design cycles with LiveDesign
Dispersed teams and remote work environments can pose a significant obstacle to real-time collaboration in a drug discovery program. LiveDesign, Schrödinger’s cloud-based enterprise informatics platform, enables project teams to design compounds, run predictive models, and analyze experimentally-generated data in one place, allowing teams to drive design-maketest- analyze (DMTA) cycles either independently or collaboratively in teams. Centralizing data access and design tools within LiveDesign was imperative to the project team’s ability to tackle their α4β7 integrin inhibitor design challenge (Figure 1).
Using LiveDesign, the Morphic and Schrödinger teams efficiently evaluated and triaged thousands of design ideas much faster than with traditional methods, with far fewer meetings required. Using LiveDesign as the data and predictive modeling platform allowed the teams to utilize a multi-parameter optimization (MPO) strategy driven by advanced data analytics, accurate biophysical modeling of potency and selectivity, and predictive models for pharmacokinetic (PK) properties. Enabled by seamless integration of data, visualization, and predictive modeling, the team rapidly prioritized molecules for synthesis and testing, which resulted in the identification of novel chemical matter that combined the key properties required to overcome all hurdles.
Figure 1: Modeling strategy and DMTA cycle employed in LiveDesign for the α4β7 inhibitor program.
Gaining potency and balancing ADMET properties through rigorous computational assays
Through generation of over 100 proprietary co-crystal structures of inhibitor-bound α4β7 as well as α4β1 — a key off-target — the project team gained significant novel insight into the structure-activity relationships (SAR) of potency and selectivity. However, balancing PK properties with potency remained unsolved. To address this challenge the team combined advanced pKa prediction (a parameter known to correlate with oral bioavailability) using quantum mechanics (Jaguar) with free energy perturbation simulations (FEP+) for accurate potency prediction. The accuracy and utility of FEP+ as a computational assay for the prediction of relative binding energies of molecules has been validated extensively, generating predictions within 1.0 kcal/mol of experimental values on average.1
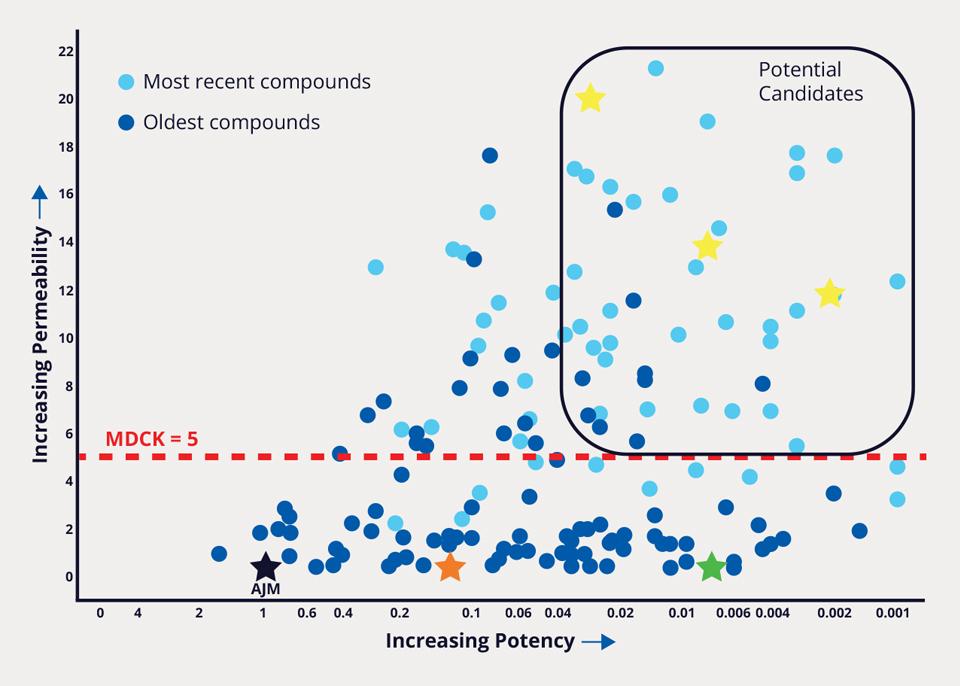
Over 8,500 FEP+ calculations were performed, enabling the team to improve potency 1000-fold, while simultaneously addressing PK liabilities. In parallel, a membrane permeability prediction model was applied to each design as part of the MPO strategy. Compounds that met the computational MPO were then synthesized in the lab. All resulting data obtained for synthesized analogs — both modeled and experimentally derived — were stored and analyzed in LiveDesign. Compounds that were identified to satisfy potency, selectivity, and permeability criteria were aggressively pursued with further characterization (Figure 2). The team’s efforts culminated in the design and delivery of MORF-057, a clinical candidate currently in Phase 2b development for treatment of ulcerative colitis (Figure 3).
Figure 2: Improving cellular potency and permeability over time using a hypothesis driven, property-focused computational design strategy. The data points in dark blue represent the early compounds, while the data points in light blue represent the more recently designed compounds. The red dashed line marks the MDCK value of 5; above 5 correlates to an increased absorption. AJM (black star) is a competitor compound that is approved in Japan, requires three doses per day, and is not selective between α4β7 and α4β1. A promising early compound designed by Morphic (orange star) was further optimized and resulted in a compound (green star) with a potency 200x greater than AJM. Additional optimization resulted in a number of potential candidates in the upper right square, including what would become MORF-057.2
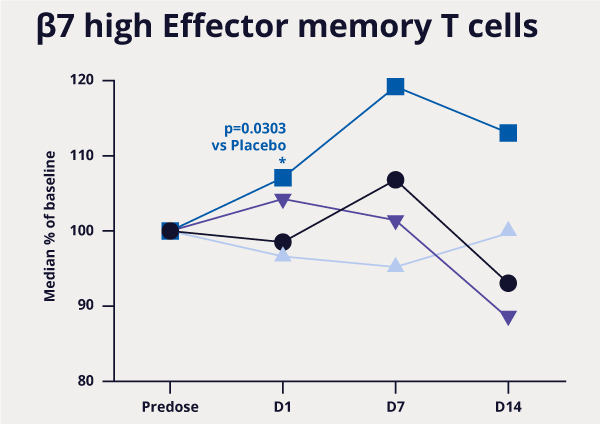
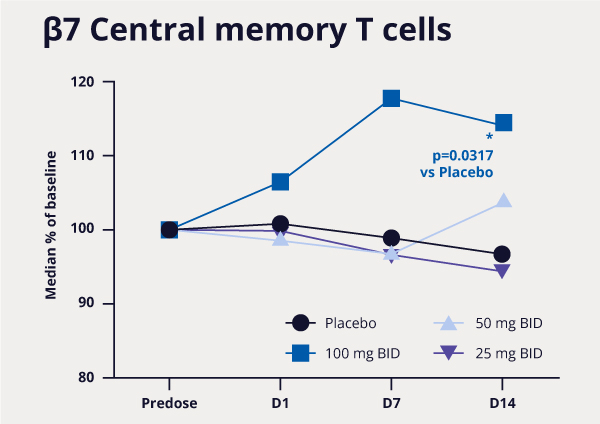
Figure 3: MORF-057 demonstrated changes in pharmacodynamic biomarkers consistent with those of approved antibody drugs.3
Enabling digital technologies to drive discovery programs
FEP+
Digital assay for predicting proteinligand binding across broad chemical space at an accuracy matching experimental methods.
Advancing drug discovery through enhanced free energy calculations.
Abel et al. Acc. Chem. Res. 2017, 50, 7, 1625–1632.
Accelerating first-in-class and best-in-class programs using a large-scale digital chemistry strategy.
Bursavich et al. Endpoints Webinar.
MORF-057, an oral selective α4β7 integrin inhibitor for Inflammatory Bowel Disease, leads to specific target engagement in a single and multiple ascending dose study in healthy subjects.
Ray et al. ECCO 2021.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Tremendous demands for development of new materials with improved performance and greener chemistry
Pressure to bring products to market quickly and cost-efficiently
Rapid need for informatics-based predictive models to extend the scale of materials optimization and discovery
Limited availability of large datasets of materials properties to effectively train models
Solutions
Amplify physics-based modeling with advanced machine learning technology and deep learning capabilities
Leverage machine learning to extend predictions over extensive chemical space unavailable to experimental methods
Develop customized descriptors for highly diverse material systems to improve the accuracy of predictions
Collaborate, manage disparate data, and share predictive models across project teams with a unified cloud-based enterprise informatics platform
With the rapid development of technology in the fields of energy, aerospace, electronics, foods and more, there are increasing demands for the design and discovery of new materials. To meet these demands, materials scientists seek innovative methods to optimize chemical properties and reduce time-to-market of better performing, more sustainable products. However, relying solely on traditional trialand- error experimental methods has become too costly and too slow, and is limited by experimental conditions. Thus the materials innovation R&D cycle has long benefited from the use of physics-based simulation engines such as quantum mechanics and molecular dynamics to help lower the cost of discovering novel chemistries, structures, morphologies, and compositions of materials for a wide array of applications and industries. In the past few years, the growth of computational power and the interest in building large datasets of materials properties has led to the growing adoption of materials informatics and machine learning-powered approaches in materials science. However, such methods are highly data intensive and suffer from an inability to extrapolate beyond the chemical space of the training model. There is a pressing need for rapid, informatics-based predictive models to extend the scale of materials optimization and discovery, extract property limits and design rules, and drive the natural synergy between physics-based modeling and machine learning methods. Additionally, with the rapid advance of machine learning methods, there is an increasing need to deploy and share accurate predictive models across research organizations for use by experts and non-experts alike. Successful R&D digitization efforts require a robust, collaborative platform for assessing predictive models for use in research.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Into the Clinic: Developing potent and selective kinase inhibitors using at-scale FEP and protein FEP: a Wee1 case study
Share
Speaker
Jennifer Knight
Director
Abstract
Free energy calculations are revolutionizing how early-stage drug discovery campaigns are undertaken. Robust free energy methods can rapidly provide on-target and off-target potency predictions to identify promising chemical matter for synthesis, inspiring further rounds of ideation and optimization. In this webinar, we discuss the discovery of novel Wee1 kinase inhibitors using a strategy that couples ligand free energy calculations with protein free energy calculations to simultaneously find promising chemical matter and de-risk for off-target liabilities. Designs with optimal potency and selectivity profiles can be rapidly identified and prioritized for synthesis, thereby accelerating drug discovery timelines.
De novo design of hole-conducting molecules for organic electronics
Panasonic and Schrödinger scientists designed over 50 novel molecules with improved hole mobility by performing large-scale density functional theory (DFT) calculations and machine learning inverse design.
Over 50 molecules identified with target performance profile
Performance Improved
Identified molecules with lower hole reorganization energy (up to 22% reduction) than the lowest one in training dataset
Highly Predictive Machine Learning (ML) Models Developed
Leveraged data based on DFT calculations of 250,000 molecules
New Insights Proposed
High quality de novo design complements molecular enumeration and virtual screening
Charge carrier mobility is one of the most important characteristics of semiconductor materials.
Applications in printed electronics demand molecules with high mobility. Despite rapid progress toward discovery of new molecules with improved mobility, challenges persist. For example, the impact of topological shape of the molecules on the magnitude of hole mobility is not well understood for optimized molecular design, and it can be extremely costly and time-consuming to synthesize and assess every candidate molecule. Atomic simulations and machine learning technologies can reveal novel insights which are inaccessible to experimental methods alone.
““With Schrödinger’s advanced simulation tools, we were able to explore millions of molecules and target tens of potential candidates within a short period of time, which is simply unfeasible with traditional approaches. This level of computational power changes the way we innovate. Both the scientific expertise and the excellence of technology Schrödinger brings to the table give us confidence in future collaborations.””
Nobuyuki N. MatsuzawaGeneral Manager of Panasonic Corporation
Approach
Scientists at Panasonic are challenged to develop novel organic semiconductor materials with higher efficiency. In order to drive innovation, Panasonic teamed up with Schrödinger for de novo design of new molecules leveraging the computational power and expertise of Schrödinger of high-throughput DFT calculations, machine learning/deep learning model building, and chemical enumeration.
Results
Scientists from Panasonic and Schrödinger performed a thorough benchmark study of three de novo methods and identified molecular structures in the heteroacene family, which may show improved carrier transport properties. 1 Schrödinger demonstrated strong large-scale computing capabilities and in-house expertise in machine learning to develop de novo methods based on knowledge and literature reports, building bayesian optimizers and reward engineering.
Conclusion
Scientists from Panasonic and Schrödinger have applied three major classes of de novo molecular design (inverse design) methods to the challenging problem of improving charge carrier mobility in materials science. They evaluated the performance of these methods via large-scale DFT calculation of hole reorganization energy. These methods present an attractive complement to molecular enumeration and virtual screening, and recent advances in deep learning for de novo design have yielded promising results for the design of novel materials.
Over 50 molecules were identified to have lower hole reorganization energy than lowest data in the training set (up to 22% reduction). We expect significant enhancement in hole mobility by the reduction of the reorganization energy in the newlydesigned molecules.
The best scoring compound was found by the JTNN method, followed by REINVENT. However, on the whole, the REINVENT method generated the best top 1,000 molecules.
Based on the findings, the scientists propose that high-quality de novo methods should optimize for compounds that “fill holes” in the space of the enumeration, generating highly targeted molecules.
References
De Novo Design of Molecules with Low Hole Reorganization Energy Based on a Quarter-Million Molecule DFT Screen
Gabriel Marques*, Karl Leswing, Tim Robertson, David Giesen, Mathew D. Halls, Alexander Goldberg, Kyle Marshall, Joshua Staker, Tsuguo Morisato, Hiroyuki Maeshima, Hideyuki Arai, Masaru Sasago, Eiji Fujii, and Nobuyuki N. Matsuzawa* J. Phys. Chem. A 2021, 125, 33, 7331–7343.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
First-principles simulation has become a reliable tool for the prediction of structures, chemical mechanisms, and reaction energetics for the fundamental steps in homogeneous catalysis. Details of reaction coordinates for competing pathways reveal a fundamental understanding of observed catalytic activity, selectivity, and specificity. Such predictive capability raises the opportunity to accelerate computational discovery and design of new single-site catalysts with enhanced properties.
However, along with the rapid technology development and materials innovation, challenges persist:
The complexity of chemical reactions and the need for associated computational research has increased as demands for innovation increase
The traditional rate of catalysts discovery is limited and unable to keep pace with demands for improved catalysts
Existing computational frameworks are manually intensive with limited scale, and require a high level of expertise and training
Cataloging and maintaining databases of novel catalysts is challenging and time-consuming
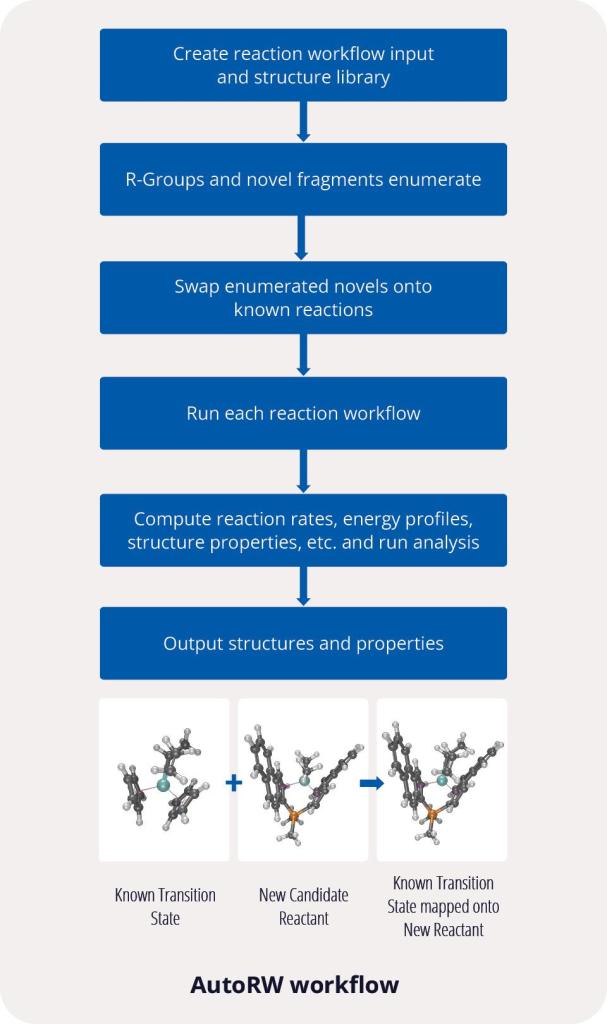
To democratize the fundamental understanding, design, and discovery of novel catalysts, Schrödinger developed an automated reaction workflow called AutoRW. AutoRW combines the elements of enumeration, mapping, organization, and output needed for high-throughput screening of catalysts, reagents, and substrates, requiring a pre-built reaction coordinate, novel chemical fragment, and any R-Groups for enumeration. By automating processes and computing the reaction coordinates, rates, energies, transition states, structures, properties, for each reaction, AutoRW streamlines the process of large-scale computational catalyst screening.
Solution: AutoRW for automated large-scale catalysts screening
Simplified, customizable workflows that enhance reproducibility and predictability
Easy to use for both expert and non-expert computational users
Increased productivity for highly-complex problems and challenges
Enhanced coverage where conformers could be missed by manual methods
Improved organization of files and properties to save time and reduce errors
Dedicated scientific & technical support and vast learning resources
Case Studies: How AutoRW Accelerates Innovation in Catalysis and Reactivity
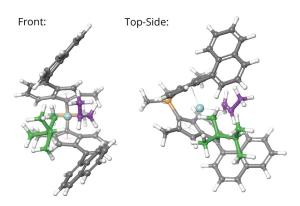
Understanding the Effects of Catalysts Selectivity on Polypropylene Tacticity
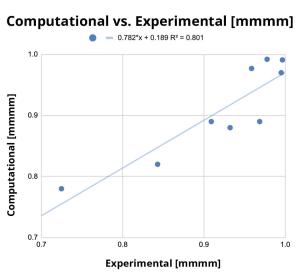
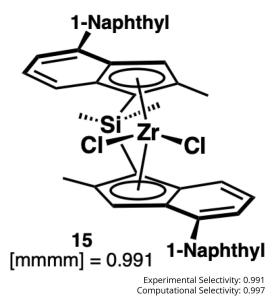
Production of olefin-based polymer products has surpassed 100 million tonnes. Of these, polypropylene is the second most produced polymer. Its physical properties are directly influenced by the regularity of adjacent stereocenters. This regularity, or tacticity, is determined by the kinetic selectivity and the control of incorporation of α-olefin monomer allows for fine-tuning of its physical properties. In this project, scientists studied 13 isotactic catalysts using AutoRW to fundamentally understand the adjacent stereoselectivity of polypropylene. The results were in good agreement with the experimental selectivities (R2 = 0.8). This quick and accurate approach allows for optimized polypropylene design and synthesis with target structures and properties.
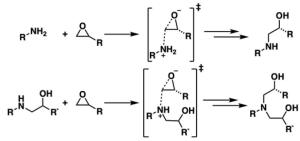
Screening Epoxy Amine Reactions for Efficient High-performance Polymer Design
Epoxy amine crosslinking reaction. The initial reaction occurs between a primary amine and a single epoxide. The resulting product contains a secondary amine that can further react with additional epoxide.
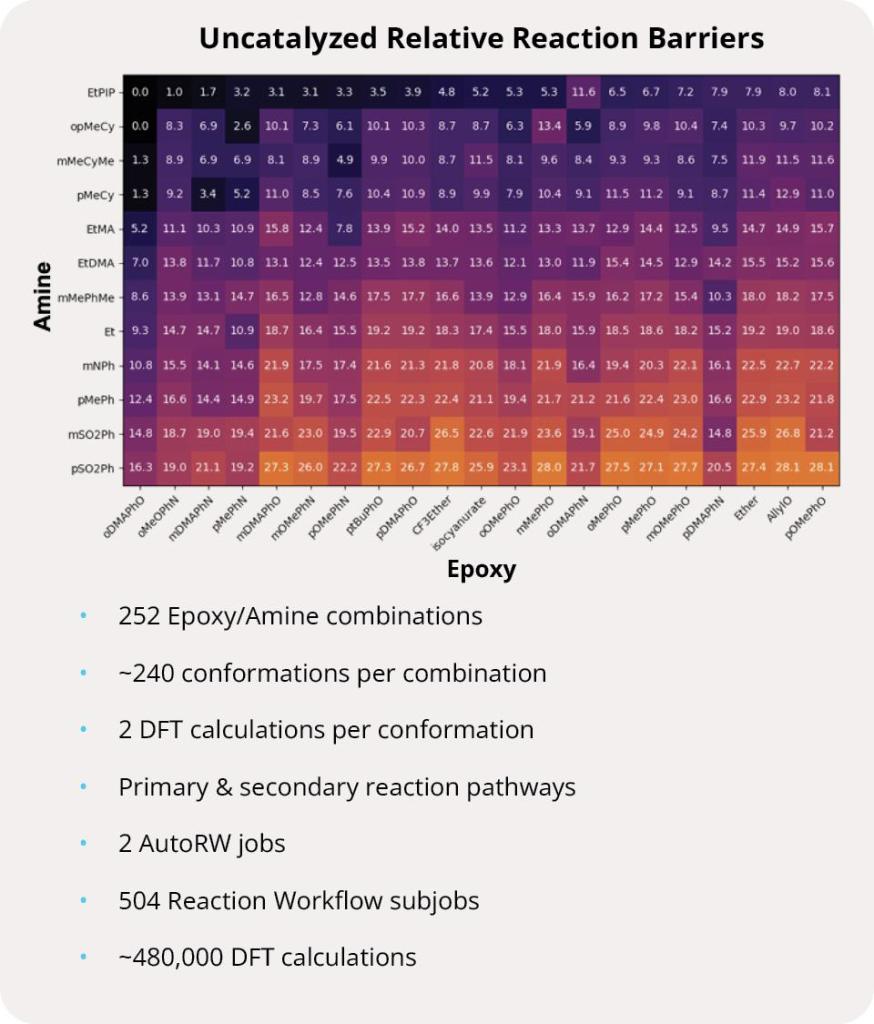
Thermoset polymers have gained interest in recent years due to their favorable thermomechanical properties for applications in aerospace, automotive, defense and high performance athletics equipment. While thermosets are very versatile, the cost to incorporate these polymers into new materials is high. Accelerating the development process pipeline would not only reduce costs, it would also decrease accumulation of thermoset waste which is difficult to recycle. Towards this end, the scientists studied a library of 12 amines and 21 epoxides to build a relative reaction barrier heat map. Each epoxy/amine combination was subjected to the Reaction Workflow to locate all stationary points in the reaction as well as compute energetic barriers for all reaction steps, enabling efficient design and synthesis of high-performance polymers.
Investigating Comonomer Selectivity for Optimized Block Copolymerization
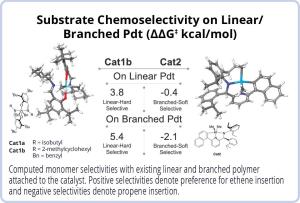
Block copolymers have unique properties that include high strength, flexibility, and melting temperature. Their physical properties are directly influenced by the polymer block length and distribution, which are then determined by the kinetic comonomer selectivity and chain shuttling agent activity. The comonomer selectivity is influenced by both the catalyst and existing polymer product. In this project, scientists ran AutoRW to screen and test 35 catalyst derivatives with different polymer substrates to understand the effect of catalysts on comonomer selectivity for different copolymerization reactions. This approach enables quick screening of catalysts and substrates to design block copolymers with target structures and properties.
Empower advanced catalysts discovery across entire R&D project teams
As the modern R&D processes evolve in a more collaborative and globalized manner, tremendous effort has been put into data storage and sharing, communication and project management across teams and geographies. Enterprise scale informatics platforms for R&D have been developed to break the silos and barriers, and have been adopted by many companies across several molecular design industries.
The benefits are clear: project teams can work across departments and sites, across geographies and time zones, and even across companies, with live sharing of all the project data of experiments, designs, processes, simulations, etc.; Teams can share, analyze and communicate data seamlessly and make rapid decisions, accelerating the collaboration and progress of the projects. Enterprise informatics platforms also simplify and optimize data management for organizations, eliminating the chaos of storing large data on computer drives and transferring data between teams and during team or personnel changes.
Schrödinger’s LiveDesign is a powerful, web-based informatics and molecular design platform that enables teams to rapidly advance materials discovery projects by collaborating, designing, experimenting, analyzing, tracking, and reporting in a centralized platform. By incorporating AutoRW into LiveDesign, scientists can get the most benefits from both.
AutoRW in LiveDesign for enterprise-wide time and cost savings
Streamlined automated enterprise solution for catalysis and reactivity
Scalable to satisfy the need of large global organizations
Integrated with advanced machine learning systems
End-to-end collaborative discovery between chemists, modelers and engineers on a single web-based platform
Live sharing of ideas and results for rapid decision-making
Intuitive cheminformatics: visualization, data and model analysis for experimental and computational data simultaneously
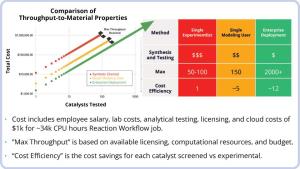
Using LiveDesign, the team can collaborate and virtually screen over 2000 catalysts per year, while a single modeling user can only screen about 150 catalysts annually. Employing the automated enterprise-scale workflows lead to much higher cost efficiency and rates of success.
Conclusions
Scientists across industries are entering a new paradigm for catalysis research. Research historically based on pure experimental trial-and-error is moving to computationally-driven workflows. Rapid technology development and evolving project collaborations demand for simplified and automated workflows at enterprise scale. Schrödinger’s AutoRW and LiveDesign enable rational catalyst design in an automated, accelerated and collaborative manner on a single web-based platform, which are easy to use and deploy across teams and organizations.
The tools empower scientists to solve high-level challenges of even more complex reactions and catalysis systems with reduced time and cost while enhancing predictability and productivity.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Advancing a Pipeline of Drug Discovery Programs Using Diverse Machine Learning Strategies Coupled with Physics-Based Simulations
Share
Speakers
Jennifer Knight, Director at Schrödinger
Karl Leswing, Executive Director, Machine Learning at Schrödinger
Abstract
The application of machine learning (ML) to drug discovery is growing rapidly, ranging from target discovery to molecular design and patient stratification. However, the lack of large, high quality datasets on which to train models limits their current utility, particularly in early discovery. Complementing ML strategies with physics-based approaches offers a way to overcome these limitations and expand the domain of applicability of ML models.
In this presentation, we describe how we are integrating machine learning strategies with physics-based modeling technologies within Schrödinger’s Therapeutics Group – from hit discovery to lead optimization and drug formulation – and the impact of these approaches on active drug discovery programs.
Key Takeaways:
Active learning strategies enable routine, efficient exploration of target-specific chemical space as well as protocol optimization to streamline project work and decisions
Automated machine learning engines within LiveDesign allow whole teams to iterate through training, reviewing and deploying models to drive the drug design and optimization process
Deep neural networks trained on QM DFT calculations (QRNN) can help guide in silico crystal polymorph prediction for de-risking drug formulation
Generative machine learning approaches can design into new, highly-targeted chemical space
Accurately predicted key thermophysical properties such as glass transition temperature (Tg), density, and coefficient of thermal expansion for the polycyanurate systems
Built realistic polycyanurate models for the first time from its constituent di(cyanate ester) monomer rather than using prebuilt triazine moieties
Effectively modeled water uptake and location in thermoset matrix by penetrant loading and radial distribution function analysis
Proposed an alternative water uptake mechanism for polycyanurate that was not observed via experimental methods
Enabled rational design of high performance thermosetting polymers
Challenges
Thermosetting polymers such as polycyanurates are important for high temperature applications in aerospace because of their performance properties, such as high glass transition temperatures (Tg) and exceptional flame resistance. Modeling and simulation efforts have enabled rapid screening of candidate materials with targeted properties for high performance applications without dedicating the significant time and resources required for wet chemistry. However, there are challenges for predicting properties of thermosetting polymers like polycyanurates:
The reactions, such as curing/crosslinking, are often complicated and require multiple factors to consider
Iterative crosslinking simulation for large cells containing thousands of reactive groups was not possible until recent years with the advances of GPU-accelerated molecular dynamics (MD) simulations
Generating a realistic model system is paramount in the prediction of properties and study of the dynamics, however, there are exceedingly few examples of in situ crosslinking of di(cyanate esters) for polycyanurate systems
Approach
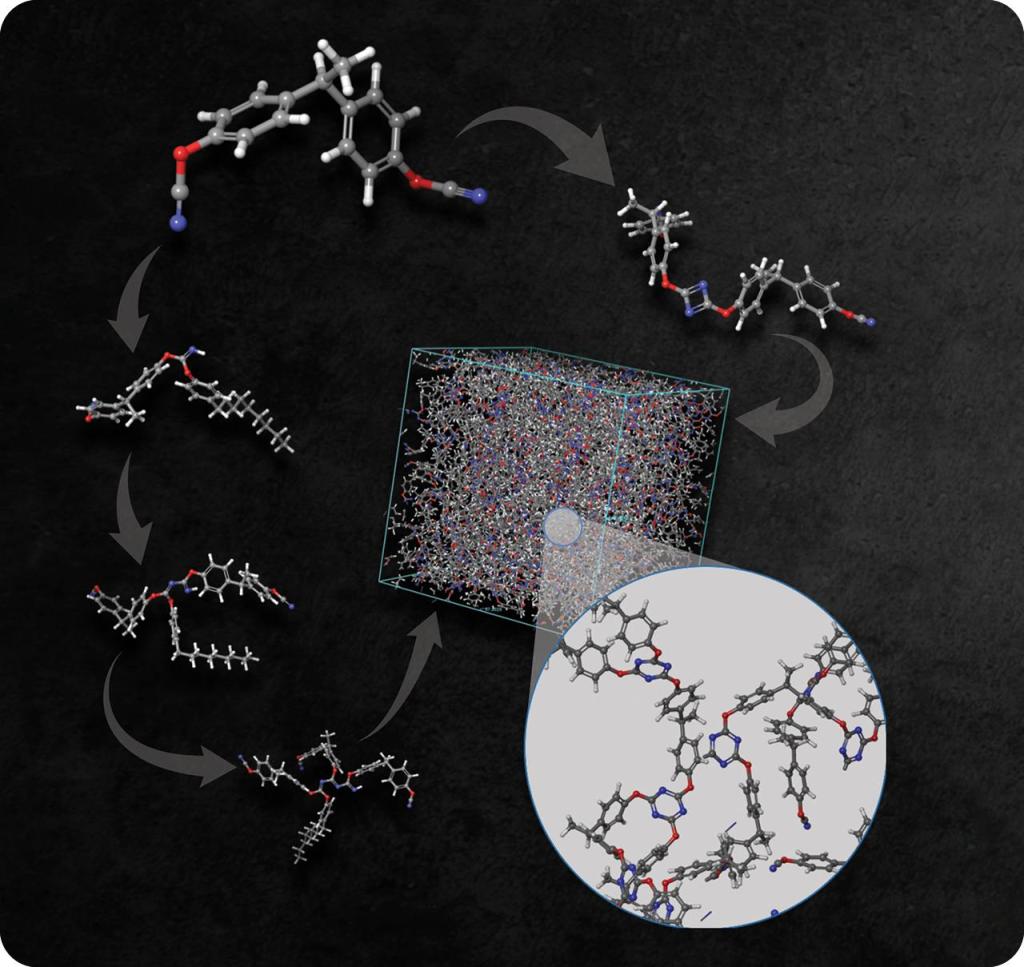
Scientists from Air Force Research Laboratory (AFRL) and Schrödinger worked together to build realistic polycyanurate models from its constituent di(cyanate ester) monomer rather than using prebuilt triazine moieties, as well as explore the crosslinking mechanisms. All chemical models were built and simulations performed using the Schrödinger Materials Science Suite and Desmond MD system:
Results
This work uses MD simulations to form polycyanurate networks in order to predict thermophysical properties and probe water uptake and aggregation behavior at the atomistic level. The predictive power of this model is considered adequate for further study of other polycyanurate systems, whether in formulation or synthesis of novel monomers to evaluate properties like water uptake or for mechanical property prediction of systems employing these and other cyanate ester materials.
Thermophysical properties (Tg, density, CTE) of the LECy systems crosslinked via the two different mechanisms aligned well with experimental data
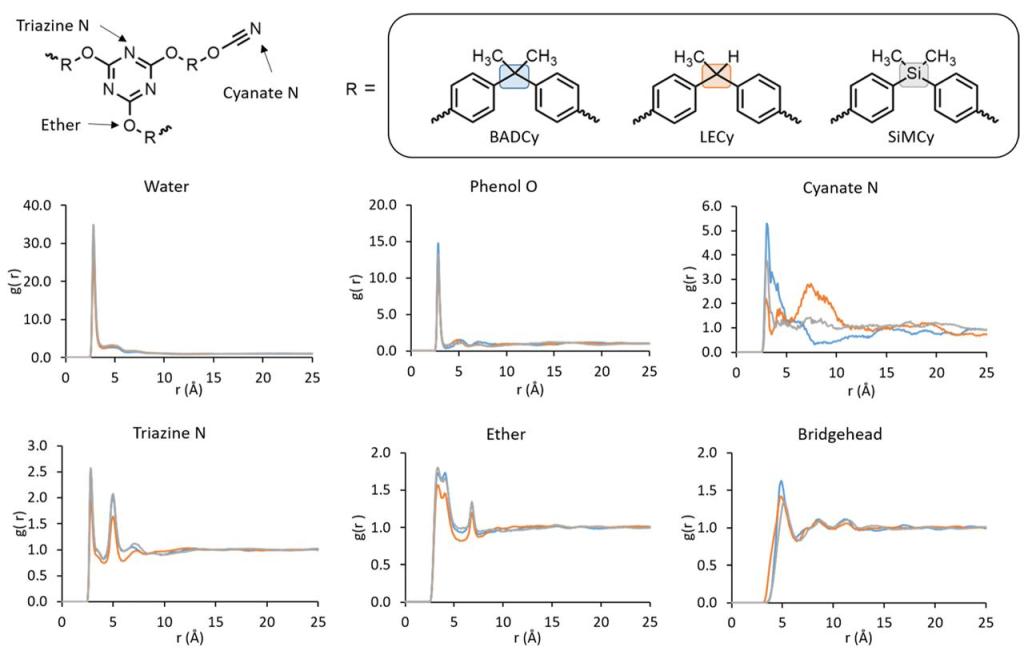
Multiple thermophysical properties were probed for BADCy and SIMCy systems, and the simulated data showed rank ordering in line with the experiment
Water overwhelmingly tended to aggregate together in free volume, rather than closely associated near specific moieties. However, polar moieties like triazine rings and phenolic groups showed an affinity to the water and tended to reside on the periphery of the free volume where the water is located
New hypothesis was proposed: Increased hydrophobicity is the driving factor in the decreased water uptake of SiMCy rather than a change in free volume in the SiMCy monomer
Representative radical distribution functions (RDFs) of water relative to the described moieties in the crosslinked networks. Curves corresponding to BADCy are in blue, LECy in orange, and SiMCy in gray. Note that the y-axes are not all at the same scale.*
References
Polycyanurates via Molecular Dynamics: In Situ Crosslinking from Di(Cyanate Ester) Resins and Model Validation through Comparison to Experiment
Levi M. J. Moore, Neil D. Redeker, Andrea R. Browning, Jeffrey M. Sanders, and Kamran B. Ghiassi, Macromolecules 2021, 54, 13, 6275–6284
*The figures were published in the referenced paper. Copyright ACS Publications.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Prediction of moisture adsorption and effects on amorphous amylose starch
Molecular Dynamics simulations accelerate optimization of product quality and processing for food and beverage, packaging, and pharmaceutical applications.
Accurately predicted key physical properties such as glass transition temperature (Tg) for both wet and dry amorphous amylose polymers
Effectively modeled water uptake and transport by investigating the effect of moisture content on Tg and water diffusion in starch polymers
OPLS3e force field provided high accuracy for amorphous starch models
Further studies proposed on in-depth water-amylose interactions and the effects of ingredients on complex starch formulation
Challenges
As amorphous starch is highly attractive for applications in thermoplastics and food encapsulation, understanding its functionality is a high priority for R&D scientists in several industries. Key to starch functionality is the interaction between water and starch granules. Numerous studies have focused on understanding the microstructure and the underlying nanostructure of starch, however a thorough understanding of the molecular structures of native and amorphous granules is challenging to characterize experimentally. This results in:
Lack of high resolution, atomic level structural characterizations
Lack of morphological or thermophysical properties of amorphous starch
Lack of dynamic properties of amorphous starch in solvent
Approach
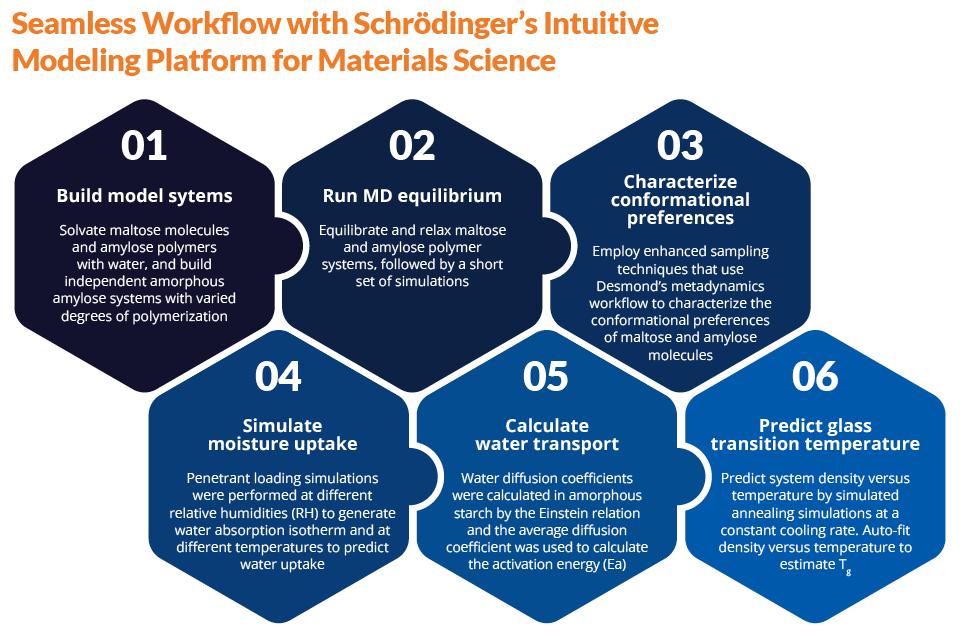
Scientists at Schrödinger aimed to solve these challenges by using molecular simulation techniques, specifically molecular dynamics (MD) simulations.1 All models and simulations were performed using the Schrödinger Materials Science Suite. These methods predicted water uptake at a given temperature and relative humidity condition and also allowed for tracking of water transport in the starch polymer matrix.
Results
MD simulations have greatly improved our understanding of amylose molecules in solution and bulk phase and have led to rapid prediction of key physical properties such as Tg.
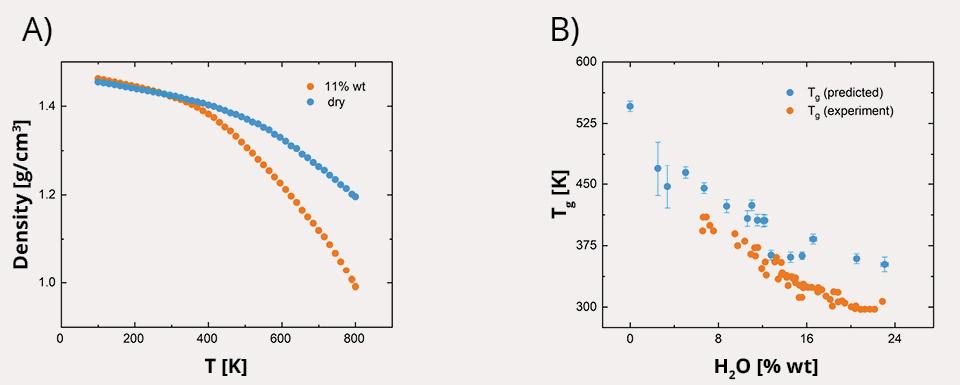
The results illustrate the plasticization effect of moisture content on amorphous starch. Predicted glass transition temperature (Tg) depression as a function of moisture content is in line with experimental trends and allows prediction of the previously inaccessible dry Tg of amorphous starch.
Using combined grand canonical Monte Carlo (GCMC)/MD simulations, a moisture sorption isotherm curve is predicted along with temperature dependence. Concentrationdependent activation energies for water transport agree quantitatively with previous experiments.
The modern OPLS3e force field and welltempered metadynamics provided highly accurate prediction of dynamical properties and conformations for amorphous starch polymers in addition to solution behavior.
Plasticization effects of water on amorphous DP31 amylose. A) Density versus temperature plots of dry and 11 % moisture content DP31 using a 0.67 ns/K cooling rate. B) Tg as a function of moisture content. Blue data points represent the average of five independent systems. Orange data points are experimental values reported in (Ai & Jane, 2018).*
What’s Next?
Further studies can explore in-depth water-amylose interactions and the effects of other commonly used ingredients in starch formulations, for example emulsifiers, which are known to bind to amylose molecules.
Future work can be performed to understand the morphology of complex amorphous and/or semi-crystalline starch formulations and how morphology affects transport and thermomechanical properties.
References
Jeffrey M.Sanders, Mayank Misra, Thomas J.L.Mustard, David J.Giesen, Teng Zhang, John Shelley, Mathew D.Halls, “Characterizing moisture uptake and plasticization effects of water on amorphous amylose starch models using molecular dynamics methods”, Carbohydrate Polymers, 252, 2021, 117161.
*The figures were published in the referenced paper. Copyright Elsevier
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.