Pathfinder-Driven Chemical Space Exploration and Multiparameter Optimization in Tandem with Glide/IFD and QSAR-Based Active Learning Approach to Prioritize Design Ideas for FEP+ Calculations of SARS-CoV-2 PLpro Inhibitors
Overview of Molecular Modelling for Formulations

DEC 19, 2022
Overview of Molecular Modelling for Formulations
Speaker
John Shelley
Fellow
Summary
This talk is an overview of molecular modeling calculations relevant for formulations in the pharmaceuticals, inks, 3D printing, polymers, batteries and agricultural chemicals industries.
Chinese: 2022薛定谔秋季中文生命科学网络讲座 | 基于物理理论的计算模拟 – 如何准确预测小分子晶体的结构和溶解度


DEC 7, 2022
2022薛定谔秋季中文生命科学网络讲座 | 基于物理理论的计算模拟 – 如何准确预测小分子晶体的结构和溶解度
Speakers
Lingle Wang
Vice President
Abstract
对固态科学家来说,药物晶体形式的改变是药物研发后期甚至上市后是非常严重的,打击性极强的问题。这类新型的晶体形式可能表现出不同的,有可能是负面的特性。许多药物的多晶型消失,包括利托那韦、罗替戈汀和盐酸雷尼替丁等,给制药公司带来了巨大的损失并引发一系列患者诉讼。本次研讨会,我将介绍一种通过大规模回顾性验证和被实际药物制剂过程中的前瞻性研究所证明的、可靠且准确的方法来预测候选药物的所有低能量稳定多晶型物及其相对稳定性。此外,我们还将重点介绍通过自由能扰动 (FEP+) 方法准确计算晶体结构的热力学溶解度。
Emergence of unexpected crystal forms during late stages of drug development or after the initial launch of the drug is a very frustrating and serious problem for solid-state scientists. Since the newly emerged forms can exhibit different and potentially undesired properties, disappearing polymorphs for many drugs, including ritonavir, rotigotine and ranitidine hydrochloride for example, have caused big losses for pharmaceutical companies and series patient litigations. In this presentation, I will present a reliable and accurate method to predict all the low energy stable polymorphs of a given drug candidate and their relative stabilities, as demonstrated both from large scale retrospective validation and prospective studies in real drug formulation processes. I will also highlight the accurate calculations of the thermodynamic solubilities of crystal structures from free energy perturbation (FEP+) method.
Chinese: 2022薛定谔秋季中文生命科学网络讲座 | 用最新的基于物理计算方法为基于结构的药物研发开辟新天地

NOV 24, 2022
2022薛定谔秋季中文生命科学网络讲座 | 用最新的基于物理计算方法为基于结构的药物研发开辟新天地
Speaker
Dr. Jianxin Duan
Fellow
Abstract
近年来,随着新的高预测性、基于物理理论方法的发展与其加速发现新型临床化合物能力的展现,基于结构的药物发现 (SBDD) 策略的价值得到提升。然而,这些方法受靶蛋白的高质量结构模型可用性的限制。 最新的结构生物学创新利器,如冷冻电镜和计算预测的蛋白质模型(使用机器学习和基于物理的方法)有望开创一个新的靶点纪元。 在本次网络研讨会中,我们将介绍最新的计算工作流程如何在这些具有历史挑战性的靶点和脱靶点上实现基于结构的药物发现。
主要议题:
在没有实验晶体结构(即同源模型或 AlphaFold 结构)的情况下,建立和验证高质量蛋白质结构模型用于SBDD的新计算方法。
通过以下案例展示新方法在项目中的作用:
1)推进用高通量筛选获得的初步苗头化合物
2)解决脱靶效应带来的障碍
3)使用同源模型推进整个项目
The value of pursuing a structure-based drug discovery (SBDD) strategy has amplified in recent years as new highly-predictive, physics-based methods have evolved and demonstrated the ability to accelerate the discovery of novel clinical compounds. However, these approaches are limited by the availability of high-quality structural models of the target protein. Recent advances in structural biology such as cryo-EM and computationally-predicted protein models (using machine learning and physics-based methods) have the potential to open a new world of targets to pursue. In this webinar, you’ll learn how new advances in computational workflows are enabling structure-based drug discovery on these historically challenging targets and off-targets.
Key topics covered:
Overview of new computational approaches for building and validating high-quality protein structural models for use in SBDD in the absence of an experimental crystal structure (i.e. homology models or AlphaFold structures)
Case studies demonstrating the impact of these approaches to:
1) progress initial hits from high-throughput screens
2) dial-out off target liabilities
3) progress entire programs using homology models
Driving innovation in polymer R&D with molecular simulation & machine learning
Driving innovation in polymer R&D with molecular simulation & machine learning
R&D scientists across broad industries from aerospace to electronics face challenges in developing the next-generation of polymers and composites with high-performance, multifunctional capabilities that also meet society’s demands for miniaturization and environmental sustainability.

Solution Overview
Chemical reactivity, physical morphology, and polymer physics drive the behavior of polymers and soft materials. Schrödinger’s Materials Science platform provides a unique combination of capabilities for the design and optimization of polymers, including:
- Differentiated model builders
- Extremely efficient, GPU-powered molecular dynamics (MD) engine
- Automated thermophysical and mechanical response workflows
- Chemically adaptable cross-linking workflows
- Analysis tools for the simulation, optimization, and discovery of novel polymers including linear, crosslinked, elastomers, dendrimers, and copolymers
Schrödinger’s tailored solutions for molecular modeling of polymers, mixtures, and composites can reduce cost and risk, shorten timelines and drive innovation in a broad range of industries.
Industries and Applications
Automotive and Aerospace
- Optimize polymer formulation properties: Model water transport, uptake and morphological stability in polymer composites using multistate simulations
- Accelerate chemical design to properties critical to product and processing constraints: Identify the unique combinations of epoxy-amine reactions for target properties using highthroughput screening
- Accelerate the manufacturing process pipeline: Predict polymer gelling during manufacturing process
- Design greener products that are more environmentally sustainable: Simulate and predict properties of high-performance resins with bio-based materials, and automate discovery of new biomaterials
CPG Packaging and Formulation
- Develop environmentally sustainable packaging: Study interfacial interactions between packaging materials and consumer goods, and simulate water uptake for barrier design and performance
- Optimize polymer formulation: Quantify the diffusion of additives in polymer packaging materials
- Minimize production waste: Understand the potential causes and impact of polymer production deviations
- Innovate with natural materials: Explore active, recyclable, and bio-based materials through molecular simulation
- Reduce processing cost: Evaluate new formulations for potential manufacturing challenges
Specialty Polymers
- Speed up decision-making on catalyst selection for raw materials production: Simulate and understand the catalysis mechanisms, selectivity, and reactivity of epoxy amine, urethane, and other reactions
- Optimize the design of high-performance polymers: Predict glass transition, thermal stability, and thermal expansion with new polymers
- Drive innovation in polymer reactions: Predict curing kinetics and processing properties
Electronic Packaging
- Identify thermal/mechanical performance issues early in design: Simulate coefficient of thermal expansion for packaging polymers
- Optimize chemical stability: Simulate interactions of packaging polymers with processing solvents and water to predict stability in use
- Design new monomers for improved performance: Predict design drivers like Dk, Df and refractive index with physics-based simulations and machine-learned models
Batteries
- Understand mechanisms at molecular level: Predict electrolyte degradation reaction mechanisms and control energetics to understand the formation of functional solid electrolyte interphase layers at anodes (SEI) and cathodes (CEI)
- Identify best-performing battery materials: Perform high-throughput screening of anode, cathode, electrolyte and additive materials to identify best chemistries for development of nextgeneration batteries
- Optimize electrolyte design: Simulate interatomic interactions and transport of ions in liquid and polymer electrolytes
Pharmaceutical Formulation
- Optimize drug carrier design: Evaluate the solubility/miscibility of drugs with different polymeric carriers or barriers
- Optimize drug formulations: Visualize and characterize pH-dependent polymeric surfactant and drug interactions in solution and precipitation inhibition behavior
- Drive effective drug delivery: Simulate drug release profiles from amorphous solid dispersions into solution
Team Collaboration and Digital Data Management
- Empower collaboration: Employ web-based enterprise informatics tools for sharing experimental and predictive models seamlessly
- Amplify research: Rapidly deploy of machine learning models to drive predictions and assist novel design approaches
- Improve project management: Accelerate project communication and collective learning by capturing, analyzing, and testing new ideas and data in a centralized platform
Products
Check on the products that enable your success in the polymer industry.
References
-
Melt-state degradation mechanism of poly (ether ketone ketone): the role of branching on crystallization and rheological behavior.
Wiggins J. et al. Polym. Degrad. Stab. 2022, 200, 109968.
-
Atomistic Molecular Dynamics in PEO/PMMA Blends Having Significantly Different Glass Transition Temperatures.
Habasaki J, Int. J. Appl. Glass Sci, doi.org/10.1111/ijag.16553.
-
Experimental Investigations of AlMg3 Components with Polyurethane and Graphene Oxide Nanosheets Composite Coatings, after Accelerated UV-Aging.
Murariu A. C. et al. Molecules 2022, 27, 1, 84.
-
Polycyanurates via Molecular Dynamics: In Situ Crosslinking from Di(Cyanate Ester) Resins and Model Validation through Comparison to Experiment.
Moore L. M. J. et al. Macromolecules 2021, 54, 13, 6275–6284.
-
Photophysical Properties of Cyclometalated Platinum(II) Diphosphine Compounds in the Solid State and in PMMA Films.
Anderson C. M. et al. ACS Omega 2021, 6, 42, 28316–28325 High-Throughput Molecular Dynamics.
-
Simulations and Validation of Thermophysical Properties of Polymers for Various Applications.
Afzal. A et al. ACS Appl. Polym. Mater. 2021, 3, 2, 620–630 Fundamental Limits to the Electrochemical Impedance Stability of Dielectric.
-
Elastomers in Bioelectronics.
Floch P. L. et al. Nano Lett. 2020, 20, 1, 224–233.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Computational chemistry applications

Computational chemistry applications
An in-depth exploration of computational chemistry applications to solve real-life biological science, materials, and engineering problems.

Computational chemistry allows researchers to explore a large, diverse range of chemical space since it is much easier to draw a molecule on the computer than to synthesize, purify, and characterize a molecule in a lab.
When deployed appropriately, computational chemistry applications can effectively bring molecules to life on the computer by accurately simulating and predicting relevant properties. For instance, the binding affinity of a small-molecule ligand to a protein target can be calculated with a similar accuracy to that of wet lab assays.
Within computational chemistry, physics-based methods grounded in first-principles can enable prediction accuracy matching experimental accuracy and are broadly applicable, but they tend to be more computationally expensive than other methods. Alternatively, machine learning (ML) methods, which develop a model by training on a data set, are also being deployed for molecular design. These ML approaches can generate results much faster but are most effective when exploring chemical space that is related to the data set the machine learning model is built upon, thus limiting their domain of applicability.
Combining physics-based and ML approaches incorporates the strengths of both to speed up scientific advances in molecular design. For example, integrating active learning into physics-based molecular docking allows one to assess very large chemical libraries in an efficient manner while still retaining the high level of performance. With active learning incorporated in docking algorithms, roughly 30,000 compounds can be tested in one second as compared to typical non-ML methods that run at roughly 1 compound per 30 seconds–this represents a 104 times speed up.
Putting Computational Chemistry to Work
Many industries are using computational chemistry methods and molecular modeling to drive innovations in pharmaceutical drugs, packaging materials, batteries, and more. Some applications for computational chemistry include:
- Drug design
- Medicinal chemistry design
- Chemoinformatics
- Consumer packaged goods
- Protein/antibody engineering
- Enzyme design
- Organic electronics
- Pharmaceutical formulations
- Catalysis design
- Polymer design
- Surface chemistry
- Energy capture and storage
- Lead optimization
- Drug target validation
- Semiconductors
- Peptide design
- Metals, alloys, and ceramics design
Benefits of Using Computational Chemistry
Computational chemistry aims to simulate and predict molecular structures and properties using different kinds of calculations based on quantum and classical physics. Advances in machine learning are also making computational chemistry more effective by increasing the speed at which calculations can be done.
Computational chemistry methods reduce the time, money, and reagent resources spent on synthesis, assays, and other experimental work. Machine learning applications can further enhance computational chemistry by increasing the speed of complex calculations, sometimes by several orders of magnitude. By carefully integrating machine learning with physics-based algorithms, digital chemical design can easily outpace wet lab design. This time savings directly translates into cost savings. Additionally, these methods allow for a broader expanse of chemical space to be explored, which can result in a greater likelihood of finding unexpected, novel molecules. In the fast-paced world of molecular design, where first-to-patent can mean the difference between success and the loss of a research program, the increase in the speed and breadth afforded by digital chemistry increases the chances of owning intellectual property.
Real-World Computational Chemistry Applications
Computational Chemistry Accelerates Drug Design
When used in drug discovery programs, computational tools allow the exploration of the chemical space with times and costs that cannot be achieved with wet-lab experiments.
For example, recent acceleration of the lead optimization process was made by using a broad search algorithm and cloud computing to explore a huge chemical space–more than 1 billion molecules computationally characterized–towards the goal of designing new inhibitors of d-amino acid oxidase (DAO). DAO is a target for the treatment of schizophrenia. This work shows the application of chemical enumeration, property filtering, machine learning and rigorous free energy perturbation calculations to design new small-molecule drugs and tackle the multiparameter optimization problem.
R&D for Product Development in Consumer Packaged Goods
In the consumer packaged goods (CPG) industry, manufacturers need to consider cost, performance and sustainability when developing new products.
Computational chemistry models and simulations decrease the development timeline and costs by allowing for fast screening, design and testing of new materials. Reckitt, which produces health, hygiene and nutrition consumer products, uses quantum mechanics and molecular dynamics computational tools in their R&D process to speed innovation. They have described how they used digital chemistry in their efforts to design more sustainable materials and how this approach has sped up timelines by 10x on average compared to a solely experimental approach.
Physics-Based Simulations to Develop New Energy Solutions
Another exciting application of computational chemistry approaches is the use of atomic-scale materials modeling in the design of new battery and energy storage solutions.
Some behaviors of materials that have been studied include ion diffusion, electrochemical response in electrodes and electrolytes, dielectric properties, mechanical response, and more. This computational approach has been used to screen for Li-ion battery additives that form a stable solid electrolyte interphase.
Driving R&D with Schrödinger’s Pioneering Computational Platform
At Schrödinger, our physics-based computational platform allows companies worldwide to harness the capabilities of computational chemistry methods and apply these to their R&D programs quickly and with ease. Over the last 30 years, Schrödinger’s modeling software and services have enabled the discovery of high-quality, novel molecules and materials across industries–as illustrated by some of the examples described above.
Molecules come to life in Maestro, the streamlined portal for structural visualization and access to cutting-edge predictive computational modeling and machine learning workflows. And researchers can bring their digital and experimental data side-by-side within LiveDesign, Schrödinger’s enterprise informatics platform for collaborative analysis, molecular design, and program management.
As the predictive and analytical capabilities of physics-based modeling continue to advance and are enhanced by the addition of new ML models, the myriad applications that are impacted by computational chemistry will continue to grow.
References
-
Advancing Drug Discovery through Enhanced Free Energy Calculations
2017. Abel R, Wang R, Harder ED, Berne BJ, and Friesner RA. Accounts of Chemical Research. 50(7):1625-1632. DOI: 10.1021/acs.accounts.7b00083
-
Docking and scoring in virtual screening for drug discovery: methods and applications
2004. Kitchen D, Decornez H, Furr J, et al. Nature Review Drug Discovery. 3:935–949. DOI: 10.1038/nrd1549
-
Efficient Exploration of Chemical Space with Docking and Deep Learning
2021. Yang Y, Yao K, Repasky MP, Leswing K, Abel R, Shoichet BK, and Jerome SV. Journal of Chemical Theory and Computation I. 17(11): 7106-7119. DOI: 10.1021/acs.jctc.1c00810
Chinese: 2022薛定谔秋季中文生命科学网络讲座 | 薛定谔计算模拟技术助力新型 D-氨基酸氧化酶抑制剂的发现
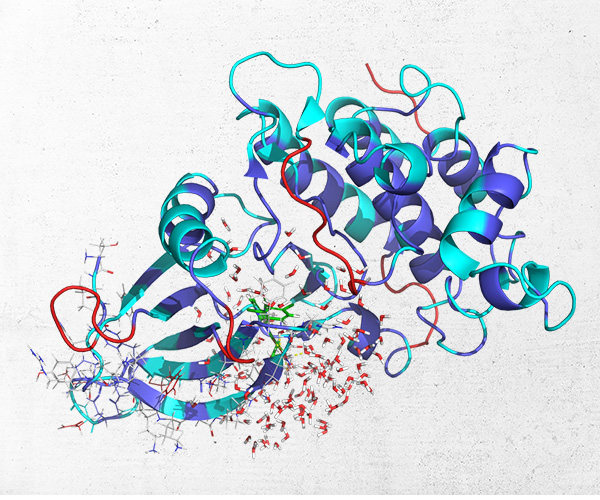

NOV 10, 2022
2022薛定谔秋季中文生命科学网络讲座 | 薛定谔计算模拟技术助力新型 D-氨基酸氧化酶抑制剂的发现
Speaker
Dr. Zhe Nie
Executive Director
Abstract
D-Serine是N-甲基d-天冬氨酸 (NMDA) 受体的共激动剂,而NMDA受体是一种关键的兴奋性神经递质受体。在大脑中,D-Serine由丝氨酸消旋酶从其L-异构体合成,并由 D-氨基酸氧化酶 (DAO, DAAO) 代谢,其中DAO是一种催化D-氨基酸(包括D-Serine)氧化降解的黄素酶, 其产物是相应的α-酮酸。许多研究已经证实了低D-Serine浓度和/或DAO高度表达以及增强酶活性与NMDA功能障碍和精神分裂症之间的关联。至此,许多公司开始探索使用DAO抑制剂治疗精神分裂症和其他适应症的可能性。我们的研究项目基于薛定谔计算建模平台的支持,以开发具有best-in-class性质的新型 DAO 抑制剂。这项研究使用hDAO FEP+模型前瞻性地预测了化合物对hDAO的抑制效力,并通过我们的AutoDesigner算法对人工设计和计算机列举的设计构思进行排序。最后,我们发现了一类具有理想药代动力学和脑渗透特性的新型DAO抑制剂。在体内小鼠 PK/PD 模型中,工具化合物37证明了通过抑制DAO功能对血浆和大脑中D-Serine浓度的调节。持续的SAR工作使DAO在生化和细胞实验中体现的效力得到了显著提高。在项目过程中,我们的建模技术不仅提高了药物化学研发的效率,还有助于识别未曾探索过的子口袋,进一步开发 SAR。
D-Serine is a co-agonist of the N-methyl D-aspartate (NMDA) receptor, a key excitatory neurotransmitter receptor. In the brain, D-Serine is synthesized from its L-isomer by serine racemase and is metabolized by the D-amino acid oxidase (DAO, DAAO), a flavoenzyme that catalyzes the oxidative degradation of D-amino acids including D-serine to the corresponding α-keto acids. Many studies have linked decreased D-serine concentration and/or increased DAO expression and enzyme activity to NMDA dysfunction and schizophrenia. Thus, many companies have explored the possibility of employing DAO inhibitors for the treatment of schizophrenia and other indications. Powered by the Schrödinger computational modeling platform, we initiated a research program to identify novel DAO inhibitors with best-in-class properties. The program execution leveraged an hDAO FEP+ model to prospectively predict compound hDAO inhibitory potency and prioritize design ideas from both human design and computer enumeration by our AutoDesigner algorithm. A novel class of DAO inhibitors with desirable pharmacokinetic and brain penetration properties were discovered from this effort. In an in vivo mouse PK/PD model, tool compound 37 demonstrated modulation of D-serine concentrations in the plasma and brain through inhibition of DAO function. Continued SAR work has led to significant potency improvement in both DAO biochemical and cell assays. Our modeling technology on this program has not only enhanced the efficiency of medicinal chemistry execution, it has also helped to identify a previously unexplored subpocket for further SAR development.
Decisive role of water and protein dynamics in residence time of p38′ MAP kinase inhibitors
Chinese: 2022薛定谔秋季中文生命科学网络讲座 | AutoDesigner,一种通过快速探索大型化学空间来优化先导化合物的从头设计算法
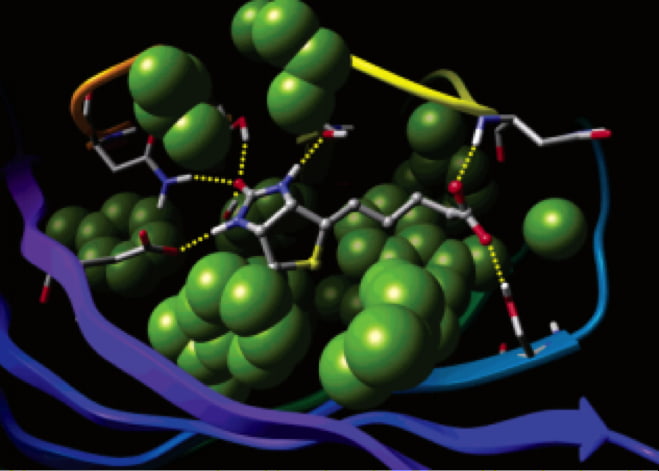

OCT 27, 2022
2022薛定谔秋季中文生命科学网络讲座 | AutoDesigner,一种通过快速探索大型化学空间来优化先导化合物的从头设计算法
Speaker
Dr. Jianxin Duan
Fellow
Abstract
药物发现中先导优化阶段通常涉及数百至数千个化合物的设计、合成和检测。设计阶段通常利用传统的药物化学方法,同时如果有合适的结构信息,也应用基于结构的药物设计(SBDD)方法。这种方式的两个主要局限性是:(1)难以快速设计出符合多个项目标准的有效分子,或解决多参数优化(MPO)问题;(2)与巨大的化学空间相比,探索的分子数量相对较少。为了解决这些限制,我们开发了AutoDesigner,一种从头设计的算法。AutoDesigner采用了云原生多阶段搜索算法,进行连续的化学空间探索和过滤。在符合项目标准范围内,比如理化性质和活性,我们可以探索和优化百万或几十亿的虚拟化合物。这算法值需要单个有活性数据和假想结合模式的小分子,非常适合早期数据贫乏的SBDD项目。
The lead optimization stage of a drug discovery program generally involves the design, synthesis, and assaying of hundreds to thousands of compounds. The design phase is usually carried out via traditional medicinal chemistry approaches and/or structure-based drug design (SBDD) when suitable structural information is available. Two of the major limitations of this approach are (1) difficulty in rapidly designing potent molecules that adhere to myriad project criteria, or the multiparameter optimization (MPO) problem, and (2) the relatively small number of molecules explored compared to the vast size of chemical space. To address these limitations we have developed AutoDesigner, a de novo design algorithm. AutoDesigner employs a cloud-native, multistage search algorithm to carry out successive rounds of chemical space exploration and filtering. Millions to billions of virtual molecules are explored and optimized while adhering to a customizable set of project criteria such as physicochemical properties and potency. Additionally, the algorithm only requires a single ligand with measurable affinity and a putative binding model as a starting point, making it amenable to the early stages of an SBDD project where limited data are available.
Antibody Humanization Guided by Computational Modeling
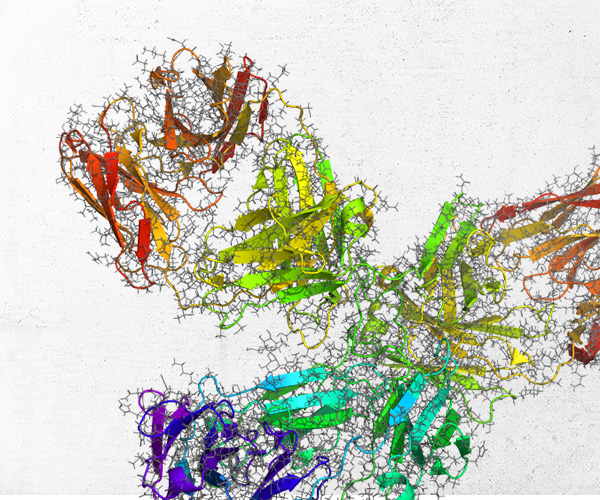

DEC 3, 2020
Antibody Humanization Guided by Computational Modeling
Speaker
Dr. Eliud Oloo
Senior Principal Scientist
Accelerating Antibody Drug Discovery Through Computational Modeling
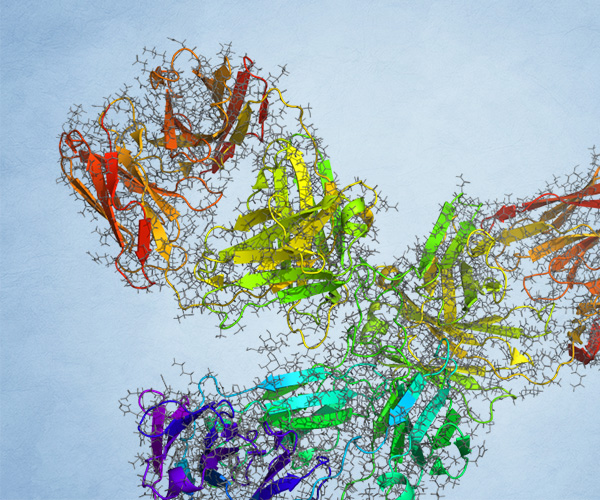

OCT 20, 2022
Accelerating Antibody Drug Discovery Through Computational Modeling
Speaker
Eliud Oloo
Senior Principal Scientist
Abstract
The large size and complexity of biologic molecules creates unique sets of safety, efficacy, and developability hurdles that have to be overcome in order to bring biotherapeutics to market. This webinar will provide an overview of computational modeling strategies for antibody design. The presentation will describe how calculated properties derived from physics-based 3D structural analyses and simulation are applied to not only predict binding affinity but also identify and mitigate potential liabilities in the development of antibody-based biotherapeutics. Such computational modeling efforts can contribute to significant reductions in project costs and timelines by directing experimental focus toward the most promising candidates.