Structure-based assessment and druggability classification of protein-protein interaction sites
A novel method for in silico assessment of Methionine oxidation risk in monoclonal antibodies: Improvement over the 2-shell model
Accelerating the design and optimization of OLED materials using active learning

Accelerating the design and optimization of OLED materials using active learning
Introduction
OLEDs (Organic Light-Emitting Diodes) are extensively used in digital displays such as those in smart phones, television screens, computer monitors, and game consoles. They contain organic molecules with unique electronic structures that create light in the visible part of the spectrum through a process called electroluminescence. For an OLED to be commercially valuable, it should satisfy several constraints, including high efficiency, low-cost fabrication, and good electrochemical stability for long-term operation, making discovery of novel OLED materials a challenging problem.
Challenges
Molecular modeling and simulation tools have proven effective in materials discovery and are increasingly deployed in industrial R&D. Although digital simulations have offered tremendous time savings for R&D workflows compared to traditional experimental approaches, several challenges persist:
- The potential chemical space for materials design and discovery is massive, even for highly constrained problems
- Predictions based on density functional theory (DFT) calculations can be laborious and computationally expensive, limiting the number of candidates evaluated within fixed timescales and resources
- It is challenging to maintain a high level of accuracy to properly assess the complexity of materials
Solution: Active Learning Workflows
A new approach is required to guide scientists to the best-performing or useful candidates. Thanks to the application of a machine learning (ML) paradigm called “active learning” (AL), Schrödinger has made this problem readily tractable. Recently, Schrödinger has developed active learning workflows which leverage the synergy between physics-based simulations and machine learning for optoelectronic properties predictions. The active learning workflow enables scientists to zoom in on the “best-performing” portion of a given sample space in a more efficient and cost-effective manner. The workflow allows:
- Automated active learning calculations with minimum users input
- Accounting for multiple optoelectronic parameters simultaneously for materials discovery
- Combining active learning with DFT to efficiently identify materials with optimal properties
- Employing built-in descriptors and fingerprints to featurize chemical structures
- Building high-performance ML models using adaptive design procedures
- Minimizing the number of time-consuming physics-based calculations
““The AL workflow enables accelerated design and discovery of optoelectronic materials. The workflow is fully automated, significantly reducing the number of physics-based simulations to predict materials properties in an extensive library. The rapid screening of datasets allows a better understanding of structure-function relationships for systematic design and application of optoelectronic materials with higher efficiency.””
Case Study
Recent studies by Schrödinger, published in Frontiers in Chemistry and presented at SID-Display Week 2022, have demonstrated the active learning paradigm for OLED materials discovery.1,2
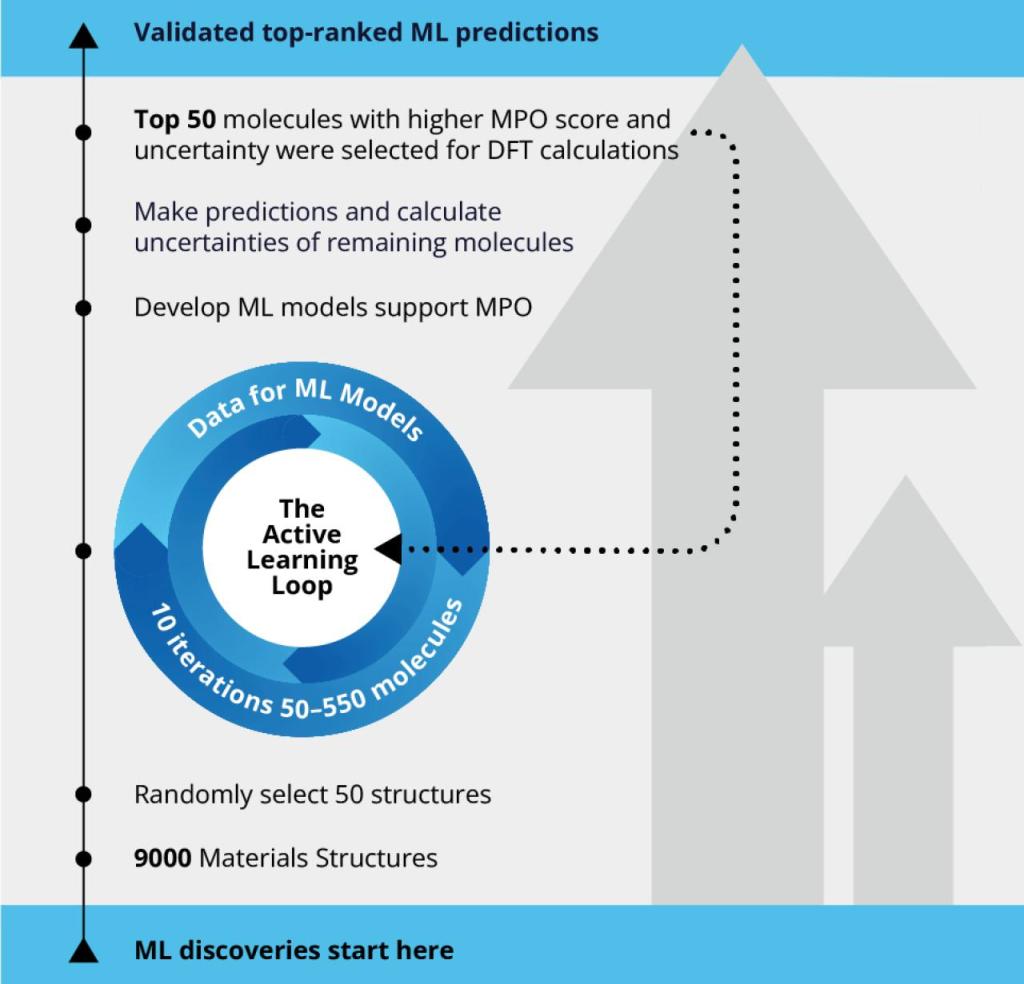
To explore the best-performing hole-transporting molecules for OLED, scientists screened a large pool of 9,000 molecules using the automated active learning workflow. The initial training set included 50 molecules for which a machine learning model was developed supporting efficient multi-parameter optimization (MPO). The ML model was then used to predict the rest of 8,950 molecules in the pool, each of which only costs a fraction of second. Top 50 molecules with higher MPO score and uncertainty were selected for DFT calculations and the calculated data were input for the next iteration of ML model training. The size of the training set for active learning was increased from 50 to 550 molecules in 10 iterations.
Active learning is the iteration of these steps until the DFT calculated data and the machine learning predicted data converge with sufficient accuracy. By using the AL approach, the scientific team was able to screen the chemical space of the materials library 18 times faster than the traditional approach of expensive quantum mechanical calculations, leading to considerable time and resource savings.
The Active Learning Workflow
References
-
Active Learning Accelerates Design and Optimization of Hole Transporting Materials for Organic Electronics Hadi Abroshan, H. Shaun Kwak, Yuling An, Christopher Brown, Anand Chandrasekaran, Paul Winget and Mathew D. Halls, Front. Chem., 2022, 9, 800371
-
Active Learning for the Design of Novel OLED Materials Hadi Abroshan, Anand Chandrasekaran, Paul Winget, Yuling An, Shaun Kwak, Christopher T. Brown, Tsuguo Morisato, and Mathew D. Halls, SID Symposium Digest of Technical Papers, 2022, 53, 885-888
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Effect of artificial sweetener saccharin on lysozyme aggregation: A combined spectroscopic and in silico approach
Exploring the effects of wetting and free fatty acid deposition on an atomistic hair fiber surface model incorporating Keratin Associated Protein 5-1
Shearing Friction Behaviour of Synthetic Polymers Compared to a Functionalized Polysaccharide on Biomimetic Surfaces: Models for the Prediction of Performance of Eco-designed Formulations
DeepautoQSAR hardware benchmark

DeepAutoQSAR hardware benchmark

Executive Summary
- This benchmark evaluates the performance of DeepAutoQSAR on two datasets of different sizes using different hardware configurations and model training times.
- Our general recommendations, based on the results and the hardware costs, are to use the NVIDIA T4 GPU hardware with the following training times: 2 hrs for datasets with less than 1,000 data points; 4 hrs for 1,000 to 10,000 data points; and 8 hrs for more than 10,000 data points.
- While performance ultimately depends on the data, the intended purpose of this benchmark is to serve as a starting point for choosing the hardware to train the ML model(s) with and the specific model training time to use. Actual performance is highly dependent on the specific dataset and may require increasing the training time or choosing a different GPU to achieve the desired results.
Introduction
The application of machine learning (ML) to predict the molecular properties of drug candidates is an important area of research that has the potential to reduce drug development timelines and accelerate the creation of medicines for patients with serious unmet medical needs.
The successful application of ML relies on sufficient data quantity and quality, a suitable model architecture(s) for the given problem, proper hyperparameter choices (the parameters for a particular ML model architecture), and appropriate model training time for a chosen hardware configuration.
DeepAutoQSAR is a machine learning product that allows users to predict molecular properties based on chemical structure. The automated supervised learning pipeline enables both novice and experienced users to create and deploy best-in-class quantitative structure activity/property relationship (QSAR/QSPR) models.
The purpose of this benchmark, which builds on the work of an earlier whitepaper [1], is to characterize the performance of DeepAutoQSAR on two datasets of different sizes using different hardware configurations and model training times. While performance ultimately depends on the data, the intended purpose of this benchmark is to serve as a starting point for choosing the hardware to train the ML model(s) with and the specific model training time to use.
Datasets
The datasets used in the benchmark were obtained from the Therapeutics Data Commons (TDC). TDC provides ML-ready datasets that can be used for learning tasks that are valuable to pharmaceutical research and development and that cover different therapeutic modalities and stages of the drug development lifecycle [2].
We use two datasets that contain assay data for one Absorption, Distribution, Metabolism, and Excretion (ADME) property each:
- Caco2 (Human Epithelial Cell Effective Permeability)
- AqSolDB (Aqueous Solubility)
Performance is measured by the median accuracy of the ADME property prediction for a sample of train-test data splits; note that the specific train-test data splits used are different from the splits provided by TDC for its benchmark leaderboard.
Dataset Descriptions
Caco2 (Human Epithelial Cell Effective Permeability) [3]*
The human colon epithelial cancer cell line, Caco-2, is used as an in vitro model to simulate the human intestinal tissue. The experimental result on the rate of drug passing through the Caco-2 cells can approximate the rate at which the drug permeates through the human intestinal tissue.
This dataset contains numerical data for use in regression, and there are 906 compounds.
AqSolDB (Aqueous Solubility) [4]*
Aqueous solubility measures a drug’s ability to dissolve in water. Poor water solubility could lead to slow drug absorptions, inadequate bioavailability and even induce toxicity. More than 40% of new chemical entities are not soluble.
This dataset contains numeric, non-integer data for use in regression, and there are 9845 compounds.
*Note: The datasets have been modified from their original form to remove structural redundancies and experimental errors.
Hardware
The hardware used in the benchmark was provisioned from the Google Cloud Platform (GCP); therefore, the hardware configurations chosen were based on the machine types offered by Google.
These limitations on hardware configurations, dictated by the cloud provider, mean that only specific hardware pairings are available, such as a particular GPU platform that can only be used with a given CPU platform. For example, NVIDIA A100 GPUs can only be run on an A2 machine type, which only uses the Intel Cascade Lake CPU platform. Constrained by these limitations, every effort was made to keep hardware-specific options consistent across machine types, to provide hardware diversity when reasonable, and to use cost-effective high-performance computing hardware.
| Hardware Key | GCP Machine Type | CPU Platform | vCPUs* | RAM (GB) | GPU Platform | GPUs | Cost ($) per Hour+ |
|---|---|---|---|---|---|---|---|
| 2 vCPUs | n2-standard-2 | Intel Ice Lake | 2 | 8 | N/A | None | $0.10 |
| 4 vCPUs | n2-standard-4 | 4 | 16 | $0.19 | |||
| 8 vCPUs | n2-standard-8 | 8 | 32 | $0.39 | |||
| 16 vCPUs | n2-standard-16 | 16 | 64 | $0.78 | |||
| T4 GPU | n1-standard-4 | Intel Ice Lake** | 4 | 15 | Nvidia T4 | 1 | $0.54 |
| V100 GPU | Nvidia V100 | $2.67 | |||||
| A100 GPU | a2-highgpu-1g | Intel Cascade Lake | 12 | 85 | Nvidia A100 | $3.67 |
** Up to Intel Ice Lake generation; GCP auto assigns CPU platform on node pool creation.
+ Prices in November 2022. Includes sustained use discounts.
Benchmarking Methods & Results
Our benchmark is a two stage process. In the first stage, DeepAutoQSAR models are trained to fit the TDC datasets using a standard cross validation procedure to select top performing ML models for the model ensemble and to optimize hyperparameters; the end result of this stage is an ensemble of top performing models, which, under normal usage, are averaged to provide a mean prediction and associated ensemble standard deviation. We detail the specific protocol in our white paper, a Benchmark Study of DeepAutoQSAR, ChemProp, and DeepPurprose on the ADMET Subset of the Therapeutic Data Commons [1]. In the second stage, random train-test splits of the data are computed, and the previously determined ensemble of top ML models architectures with specific hyperparameter configurations are trained on the new training data splits. Predictions are then generated for the new test data splits. These multi-split metrics provide a more robust estimate of model performance by reducing potential bias introduced from a single train-test data split. Model performance in this hardware benchmark is reported as the median R2 coefficient of determination [5] across these random train-test splits for each hardware configuration and model training time.
In the first stage, the initial training procedure runs continuously for each training time allotment. Due to the stochastic nature of hyperparameter optimization and model architecture selection, each hardware and training time combination can potentially explore a different number of model architectures and hyperparameter combinations each time a benchmark job is run. The model training times evaluated were: 0.5, 1, 2, 4, 8, and 16 hours. As a general rule, more competent hardware running for longer training times on smaller datasets (e.g., a machine with an A100 GPU training for 16 hrs on the smaller Caco2 permeability dataset) will explore more hyperparameterizations than less competent hardware running for shorter training times on larger datasets (e.g., a two core machine training for 2 hrs on the larger AqSolDB dataset).
Since model architecture selection and hyperparameter sampling is a stochastic process, we run each benchmark configuration, which is the particular hardware and training time combination, three times and report averages for performance—this is especially relevant when fewer hyperparameter combinations are explored as model performance is more sensitive to hyperparameter sampling. The output of the first stage is an ensemble of top models, determined by cross validation, with specific hyperparameters choices for each.
The second stage of our benchmark runs for half the training time of the first stage. Increasing training time leads to more robust statistics as the median performance converges to a split-independent value, but comes at the expense of increased computational cost; in practice computational expense must be balanced with the need to train the ensemble model for a sufficiently large training time. For performance reporting we provide the median R2 coefficient of determination [5] as computed from the multiple train-test splits, which aims to reduce potential bias introduced by a single train-test split. To compute this R2, we repeatedly split the data into training and testing sets via bootstrap sampling with replacement; to do so, we take N samples with replacement from the dataset with N total data points and remove any duplicates to form a subset. The selected points are then used to train the specific model architectures found in stage one, and the unselected points serve as the test holdout. We do this until the time limit is reached and report the median R2 of all resamplings.
As both of the TDC datasets are numerical regression problems, this metric is a reasonable measure of model performance; however, the choice of performance metric in real-world applications should always be determined according to the use-case of the ensemble model. Sometimes MAE or RMSE are more appropriate to assess if a model is sufficiently performant. The output of the second stage is a distribution of ensemble model performances over different train-test splits; the reported value is the median of the distribution.
We plot the benchmark results, which is the median R2 coefficient of determination from the second stage, below. Our first plot shows performance on the AqSolDB dataset, and the second plot shows performance on the Caco2 permeability dataset. For each of these datasets, we highlight the progression of performance over time grouped by hardware type, where hardware type is on the x-axis, training time in hours is the bar color, and median R2 score is on the y-axis. The data used to generate the plots are provided in the supplementary tables.
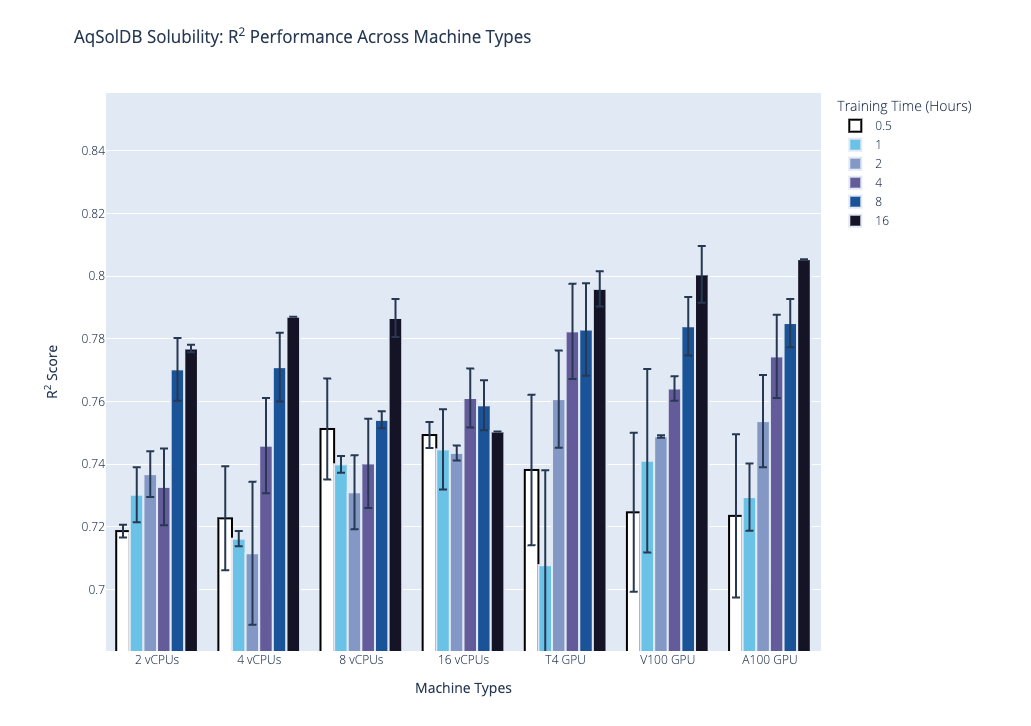
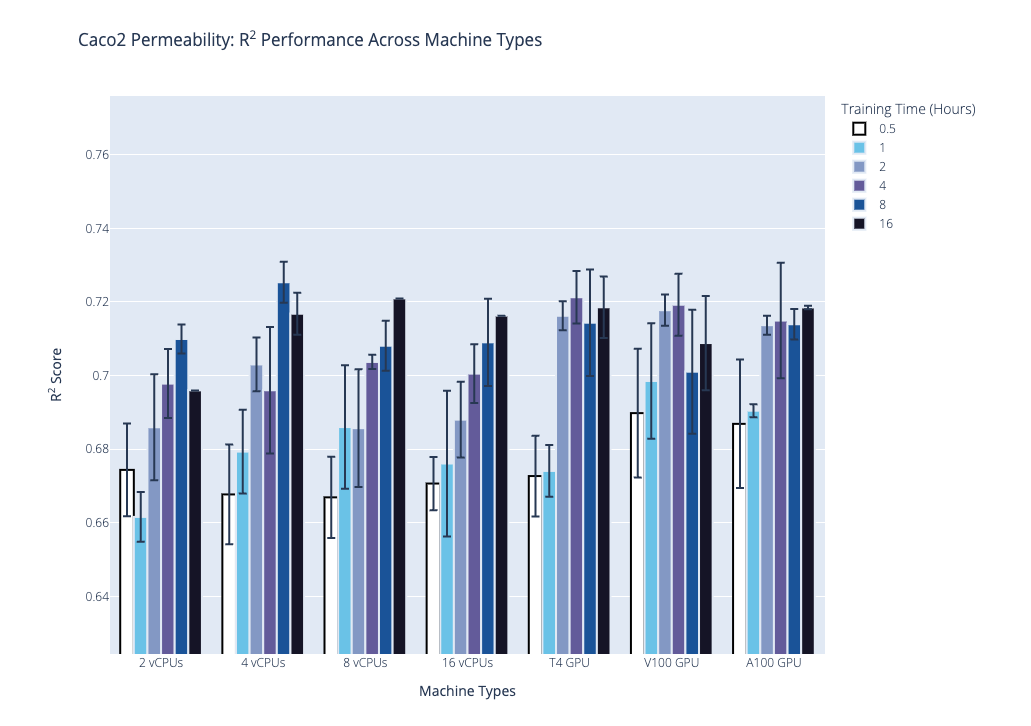
Based on these results and the hardware costs, our general recommendations are the following:
| Number of Data Points | Hardware | Training Time (hr) |
|---|---|---|
| <1,000 | Nvidia T4 GPU | 2 |
| 1,000 – 10,000 | 4 | |
| >10,000 | 8 |
Selected publications
-
Kaplan, Z.; Ehrlich, S.; Leswing, K. Benchmark study of DeepAutoQSAR, ChemProp, and DeepPurpose on the ADMET subset of the Therapeutic Data Commons. Schrödinger, Inc., 2022.
https://www.schrodinger.com/science-articles/benchmark-study-deepautoqsar-chemprop-and-deeppurpose-admet-subset-therapeutic-data (accessed 2022-11-29).
-
Therapeutics Data Commons.
https://tdcommons.ai/ (accessed 2022-06-15).
-
ADME – TDC.
https://tdcommons.ai/single_pred_tasks/adme/#caco-2-cell-effective-permeability-wang-et-al (accessed 2022-06-15).
-
ADME – TDC.
https://tdcommons.ai/single_pred_tasks/adme/#solubility-aqsoldb (accessed 2022-06-15).
-
Sklearn.metrics.r2_score — scikit-learn 1.1.3 documentation.
https://scikit-learn.org/stable/modules/generated/sklearn.metrics.r2_score.html#sklearn-metrics-r2-score (accessed 2022-11-29).
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
The role of digital chemistry across the polymer supply chain

The role of digital chemistry across the polymer supply chain
Molecular modeling and simulation tools have proven effective in materials development and are increasing in use throughout the polymer industry, from raw materials suppliers to end product manufacturers. Computational workflows also open new avenues for developing polymers with improved recyclability. Physics-based simulations offer reliable predictions of structures, morphologies, properties, and chemical reactivity for polymers. Recent advances in machine learning, deep learning, and enterprise informatics platforms have accelerated the speed, accuracy, and automation of novel materials and solutions discovery. A paradigm shift to computer-driven molecular design is occurring throughout the industry.

Vision of a digital chemistry catalyzed polymer supply chain:
Raw Materials Suppliers: Employ atomistic simulations to improve understanding and predict properties of downstream products; offer optimized raw materials to compounders.
Polymer Compounders: Use atomistic simulations to understand formulation chemistry and predict properties; provide detailed requirements to raw materials suppliers and offer optimized products to end product manufacturers.
End Product Manufacturers: Leverage atomistic simulations to predict the performance of final products and quickly identify causes of failure; give specific formulation requirements to compounders.

Solution Overview
Schrödinger’s Materials Science platform offers tailored solutions for research and business throughout the polymer supply chain, with differentiated model builders, efficient simulation engines accelerated by GPU computing power, automated thermophysical and mechanical response workflows, and accurate analysis tools.
- Broad molecular simulation and property prediction tools for: Thermal Properties, Mechanical and Dielectric Properties, Reactivity and Kinetics, Aggregation in Polymer Production, Solvent Sensitivity, Gas, Ion, Additive Diffusivity, Phase Morphology and SAXS Scattering, Semi-crystalline Morphology
- Applicable to all polymer types: thermoplastic homo and copolymers, crosslinked, elastomers, and dendrimers
- Intuitive user interface with automated workflows for experts or non-experts
- Dedicated scientific/technical support and vast learning resources
Digital Chemistry Value Across Polymer Supply Chain (Example: Transportation Industry)
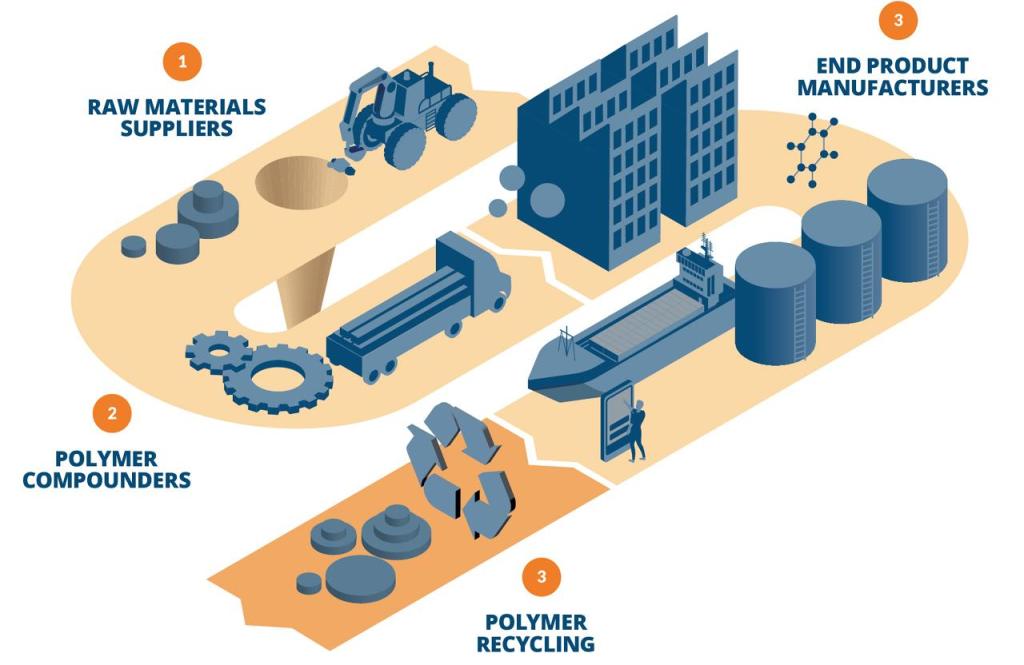
1. Raw Materials Suppliers
Suppliers of petrochemical and chemical feedstocks, additives, and various monomers and resins
Design new chemistries from alternative sources and discover new applications through simulating downstream products properties
- Predict polymer crosslinker performance in composite matrix resins such as epoxy-amine and cyanate esters
- Simulate the interaction between thermoplastic styrene-butadiene and crosslinkers
Speed decision making for catalyst selection in raw materials production
- Simulate and understand the catalysis mechanisms, selectivity, and reactivity of epoxy amine, urethane, and other reactions
Develop alternative greener raw materials that are more environmentally sustainable
- Simulate the impact of degradation on modulus for a chemistry of focus
2. Polymer Compounders
Suppliers who prepare polymer formulations by mixing or/and blending polymers and additives into process-ready products
Predict the performance of alternative raw materials in formulations and end products
- Predict glass transition, thermal stability, and thermal expansion with new polymers
- Quantify the diffusion of additive in polymers • Understand water transport and morphological stability of polymer formulations
Efficiently optimize formulation properties
- Predict and track water uptake in polymer composites
- Predict curing kinetics and processing properties
Develop greener formulations that are more environmentally sustainable
- Simulate and screen for optimal formulation with new bio-based chemistry
3. End Product Manufacturers
Processors of resins/formulations who make them into finished products on the market
Enable reliable decision-making through predictive modeling of end product properties
- Predict tire materials performance with different additives and cross-linkers
Obtain best chemistry from upstream suppliers by targeted chemical design to properties critical to product and processing constraints
- High-throughput screening of epoxy-amine reactions to identify the unique combinations for target properties
Accelerate the manufacturing process pipeline
- Predict polymer gelling during manufacturing process
Quickly screen and identify potential causes and impacts of manufacturing and material source deviations
- Predict sensitivity of matrix to cleaning solvents
Design greener products that are more environmentally sustainable
- Simulate and predict properties of high-performance resins with bio-based materials and automate discovery of new biomaterials
4. Polymer Recycling
Research and design for recyclability throughout the polymer supply chain
Design polymers for recyclability
- Predict selectivity of chemical recycling reaction
Expand use of recycled materials
- Simulate impact of recycled polymers in packaging
Determine impact on product with use of recycled material
- Screen for property changes with recycling driven microstructure changes
About Schrödinger
Schrödinger is transforming the way materials are discovered. Schrödinger has pioneered a physics-based software platform that enables discovery of high-quality, novel molecules for materials applications more rapidly and at lower cost compared to traditional methods.
Learn how digital chemistry is driving innovation across materials science industries
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Innovation in atomic-level processing with atomistic simulation and machine learning
Innovation in atomic-level processing with atomistic simulation and machine learning
Background
The Schrödinger Materials Science Suite offers computational tools specifically tailored to studying the gas-surface chemistries of atomic layer deposition and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (usually with density functional theory, DFT) and prediction of key properties. This modeling approach thus has a unique role to play both in deepening our understanding of existing processes and in discovering novel chemicals.
Many of today’s high-tech devices are manufactured by processing materials at the nanoscale, including computer chips, data storage and communications devices, sensors, solar cells and batteries. Making devices smaller, more powerful and more energy-efficient means developing new patterning, deposition and etch techniques at ever-finer resolution, in some cases down to just a few atoms thick. Chemical processing is being pushed to its limits in terms of purity, uniformity, conformality and substrate-selectivity.
Optimizing a deposition or etch process to more stringent tolerances requires a deep understanding of the underlying chemistry. Integrating new materials into a device often requires the discovery of new chemicals with improved properties. In both respects, researchers are increasingly turning to computer simulations to cut down on lab work and make discoveries faster.
The Schrödinger Materials Science Platform offers computational tools specifically tailored to tackling this problem, by studying the gas-surface chemistries of deposition, etch and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (with density functional theory, DFT) and prediction of key properties. This modeling approach can both deepen our understanding of existing processes and help discover novel chemicals.
Automate Precursor Screening for Improved Properties
Organometallic complexes can be used to deposit or etch pure metals or compounds such as oxides or nitrides. However, synthesis of the complexes in the lab can be time-consuming and difficult because many complexes react violently in air. Subsequent characterization and testing in a reactor is even more costly. Digital chemistry can rapidly screen larger numbers of chemicals than can ever be experimentally tested, narrowing down the chemical space and discovering the most promising candidate molecules for deposition or etch.
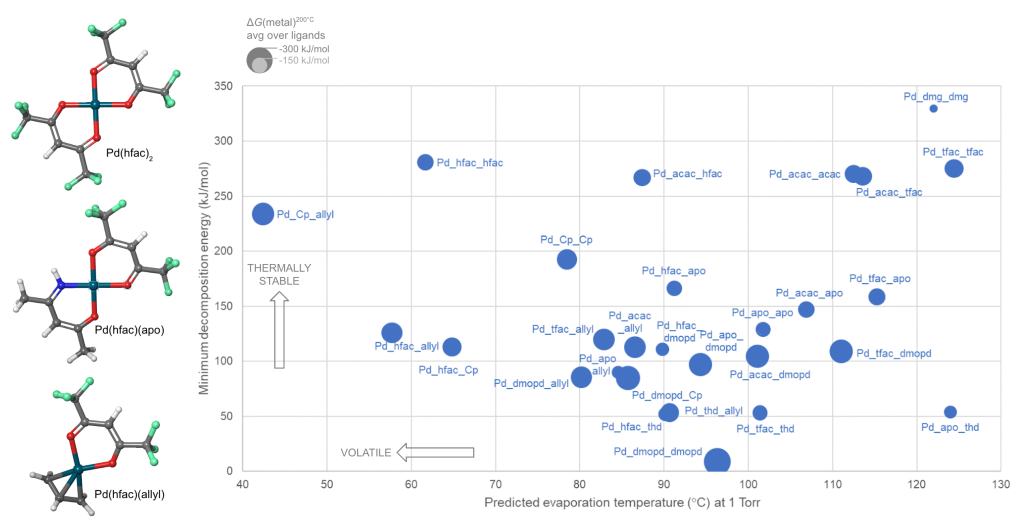
Schrödinger provides easy-to-use software for automated generation and assessment of hundreds or thousands of precursor gases. A library of hundreds of commonly-used ligands is provided, including bidentate and haptic ligands, and the library can be customized. A selection from the library is then used to enumerate and build all possible precursor complexes. We present here a case study in which a library of 10 ligands form 55 distinct complexes with divalent palladium (Pd) as the metal cation and some of these candidate complexes are shown in Figure 1.
Synthesizability
Combining two or more different ligands in a heteroleptic complex is a promising way to tune precursor properties, but it is difficult to guess whether a given combination can in fact be synthesized. This question can be answered by computing the energy for ligand exchange back to homoleptic complexes. In the Pd example, this reveals that 14 of the heteroleptic complexes are unstable and can not be synthesized.
Reactivity
Precursors for deposition must be carefully designed so as to react cleanly and deposit the desired solid material, via thermolysis or plasma activation (chemical vapor deposition, CVD) or via reactions with adsorbed fragments (atomic layer deposition, ALD). Likewise, reagents for etching must react with surface atoms to form gaseous products. Lists of candidate precursor molecules can be efficiently screened against deposition or etch chemistries using Jaguar, Schrödinger’s code for molecular DFT. Figure 1 displays the computed reactivity of the complexes with respect to Pd deposition.
Volatility and Thermal Stability
To be useful in a real CVD or ALD process, a precursor must also be volatile and thermally stable during storage and delivery. This limits the process to a window of viable temperatures, with insufficient vapor pressure at lower temperatures and impurities from decomposition at higher temperatures. For instance, Pd(thd)2 suffers from decomposition when heated to 200-250°C for the ALD of Pd metal.1, 2 Calculating volatility and thermal stability with Schrödinger’s tools gives information on how to tune these properties and widen the temperature window for maximum process flexibility.
Schrödinger has used machine learning (ML) to develop a unique model for predicting the evaporation or sublimation temperature of organometallic complexes at a given pressure. The ML-volatility model is accurate to an average of ±9°C over the training set and can evaluate hundreds of molecules per second.
The thermal stability of precursor gases can be quantified by considering the unimolecular decomposition channels: bond dissociation and β-H elimination. All such reactions in each molecule are automatically computed at the DFT level and the lowest energy reaction is selected as the most likely. The minimum decomposition energy of each complex is thus a measure of its thermal stability.
These approaches were applied to compute the properties of the Pd complexes, and Figure 1 shows how this allows the candidate precursors to be assessed.
Rational innovation is facilitated by Schrödinger’s atomistic modeling tools. Novelty is maximized by using a large library of ligands to build a wide variety of complexes, homoleptic and heteroleptic. Risk is minimized by selecting for lab study only those precursors computed to properly combine the desired properties of synthesizability, volatility, reactivity and stability.
Gain Insights Into Competing Processes for Optimum Deposition Conditions
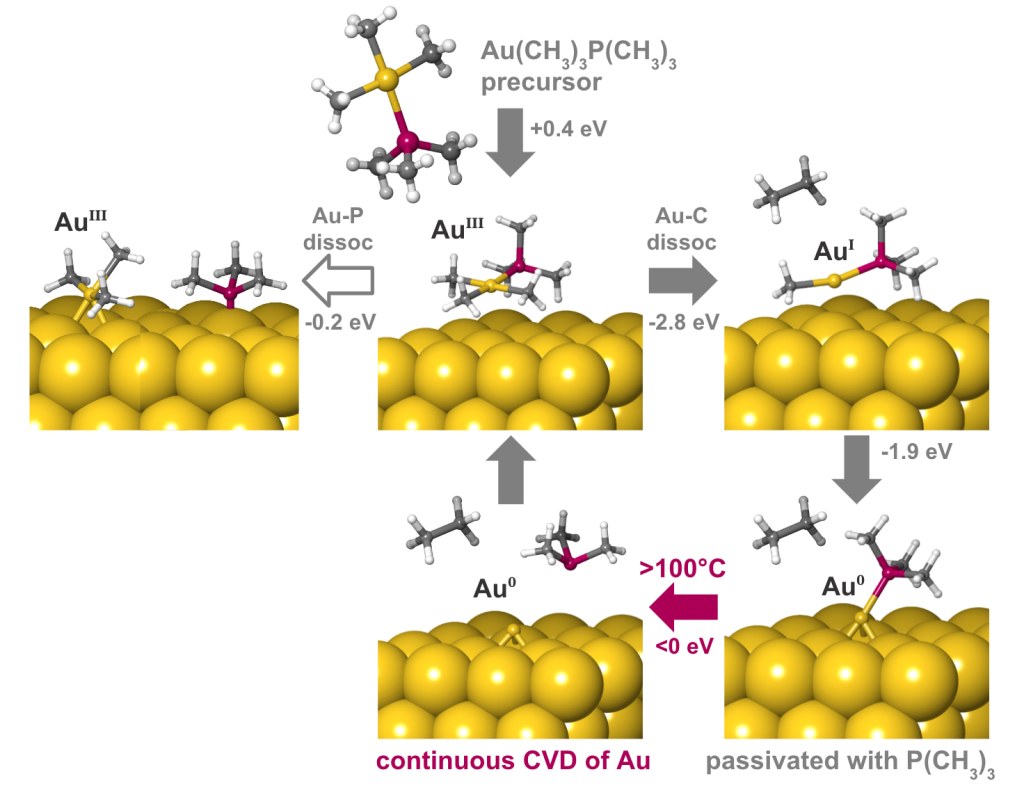
A deposition or etch process consists of a sequence of individual reaction steps, often in competition with other reactions. Simulations give atomic-level insight into these complicated mechanisms and how they depend on reactor conditions such as temperature and pressure. Here we investigate the mechanism of gold deposition from the single-source precursor trimethylphosphinotrimethylgold(III). Experiments show that gold films grow in a self-limiting fashion when this precursor is pulsed at low temperature, switching over to continuous CVD at 100°C and higher, but the reason is unknown.3
To explain this behavior, the Quantum ESPRESSO code for periodic DFT has been used to examine the detailed mechanism of reactive adsorption of the precursor onto a metallic gold surface. Thermodynamics of competing reaction steps have been computed as a function of process temperature and pressure with Schrödinger’s tools for adsorption and surface chemistry [4].
The results are summarized in Figure 2. The physisorbed precursor is in a metastable state, from which dissociation of Au-P or Au-C bonds can lead to chemisorption. Au-C dissociation and elimination of C2H6 is found to be the favored pathway, due to a strong thermodynamic driving force towards reduction of Au(III) to Au(I) and eventually Au(0). Trimethylphosphine is computed to bind to the surface, forming an adlayer that passivates against further precursor adsorption. This explains the low rate of self-limited deposition at <100°C. The computed free energies also reveal that desorption of trimethylphosphine becomes favored at higher temperatures, in agreement with the crossover to CVD seen experimentally at >100°C.
Finding out which bond breaks and how the surface is passivated opens up chemically-informed strategies for improving the process, for example by choosing a suitable co-reagent or modifying the precursor structure. Digital chemistry allows rational design to take the place of trial-and-error.
This example illustrates how changes in process behavior can be traced back to competing chemical pathways. DFT simulations give unique qualitative insight into atomic structure, bonding and oxidation states of the intermediates along each reaction pathway. In addition, quantitative information about kinetics and thermodynamics as a function of process temperature can be computed, giving valuable guidance on how to optimize the nanofabrication process. The result is improved control of purity, uniformity, phase and substrate-selectivity.
Conclusion
These cases illustrate how Schrödinger’s Materials Science Platform provides powerful and customizable computational solutions to accurately model surface reactivity, predict precursor properties and discover novel chemicals for optimized materials deposition or etch processes.
References
-
Low temperature atomic layer deposition of noble metals using ozone and molecular hydrogen as reactants,
J. Hämäläinen et al., Thin Solid Films 2013, 531, 243-250, DOI 10.1016/j.tsf.2013.01.091.
-
Atomic layer deposition of noble metals: Exploration of the low limit of the deposition temperature,
T. Aaltonen et al., J. Mater. Res. 2004, 19, 3353–3358, DOI 10.1557/JMR.2004.0426.
-
Reaction mechanism of the Me3AuPMe3–H2 plasma-enhanced ALD process, M. Van Daele et al.,
Phys. Chem. Chem. Phys. 2020, 22, 11903-11914, DOI 10.1039/C9CP06855D.
-
First-principles investigation of crossover between ALD and CVD in the thin film deposition of gold,
C. N. Brock. D. J. Giesen, A. Fonari & S. D. Elliott, Materials Research Society Spring Meeting, 2022.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.