Computational chemistry applications
An in-depth exploration of computational chemistry applications to solve real-life biological science, materials, and engineering problems.

Computational chemistry allows researchers to explore a large, diverse range of chemical space since it is much easier to draw a molecule on the computer than to synthesize, purify, and characterize a molecule in a lab.
When deployed appropriately, computational chemistry applications can effectively bring molecules to life on the computer by accurately simulating and predicting relevant properties. For instance, the binding affinity of a small-molecule ligand to a protein target can be calculated with a similar accuracy to that of wet lab assays.
Within computational chemistry, physics-based methods grounded in first-principles can enable prediction accuracy matching experimental accuracy and are broadly applicable, but they tend to be more computationally expensive than other methods. Alternatively, machine learning (ML) methods, which develop a model by training on a data set, are also being deployed for molecular design. These ML approaches can generate results much faster but are most effective when exploring chemical space that is related to the data set the machine learning model is built upon, thus limiting their domain of applicability.
Combining physics-based and ML approaches incorporates the strengths of both to speed up scientific advances in molecular design. For example, integrating active learning into physics-based molecular docking allows one to assess very large chemical libraries in an efficient manner while still retaining the high level of performance. With active learning incorporated in docking algorithms, roughly 30,000 compounds can be tested in one second as compared to typical non-ML methods that run at roughly 1 compound per 30 seconds–this represents a 104 times speed up.
Putting Computational Chemistry to Work
Many industries are using computational chemistry methods and molecular modeling to drive innovations in pharmaceutical drugs, packaging materials, batteries, and more. Some applications for computational chemistry include:
- Drug design
- Medicinal chemistry design
- Chemoinformatics
- Consumer packaged goods
- Protein/antibody engineering
- Enzyme design
- Organic electronics
- Pharmaceutical formulations
- Catalysis design
- Polymer design
- Surface chemistry
- Energy capture and storage
- Lead optimization
- Drug target validation
- Semiconductors
- Peptide design
- Metals, alloys, and ceramics design
Benefits of Using Computational Chemistry
Computational chemistry aims to simulate and predict molecular structures and properties using different kinds of calculations based on quantum and classical physics. Advances in machine learning are also making computational chemistry more effective by increasing the speed at which calculations can be done.
Computational chemistry methods reduce the time, money, and reagent resources spent on synthesis, assays, and other experimental work. Machine learning applications can further enhance computational chemistry by increasing the speed of complex calculations, sometimes by several orders of magnitude. By carefully integrating machine learning with physics-based algorithms, digital chemical design can easily outpace wet lab design. This time savings directly translates into cost savings. Additionally, these methods allow for a broader expanse of chemical space to be explored, which can result in a greater likelihood of finding unexpected, novel molecules. In the fast-paced world of molecular design, where first-to-patent can mean the difference between success and the loss of a research program, the increase in the speed and breadth afforded by digital chemistry increases the chances of owning intellectual property.
Real-World Computational Chemistry Applications
Computational Chemistry Accelerates Drug Design
When used in drug discovery programs, computational tools allow the exploration of the chemical space with times and costs that cannot be achieved with wet-lab experiments.
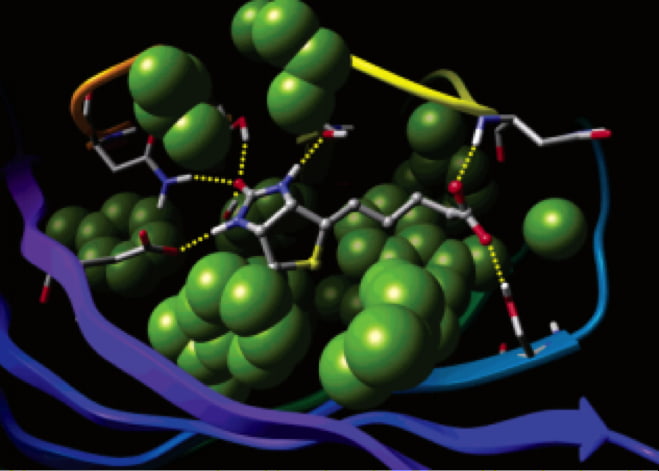
For example, recent acceleration of the lead optimization process was made by using a broad search algorithm and cloud computing to explore a huge chemical space–more than 1 billion molecules computationally characterized–towards the goal of designing new inhibitors of d-amino acid oxidase (DAO). DAO is a target for the treatment of schizophrenia. This work shows the application of chemical enumeration, property filtering, machine learning and rigorous free energy perturbation calculations to design new small-molecule drugs and tackle the multiparameter optimization problem.
R&D for Product Development in Consumer Packaged Goods
In the consumer packaged goods (CPG) industry, manufacturers need to consider cost, performance and sustainability when developing new products.
Computational chemistry models and simulations decrease the development timeline and costs by allowing for fast screening, design and testing of new materials. Reckitt, which produces health, hygiene and nutrition consumer products, uses quantum mechanics and molecular dynamics computational tools in their R&D process to speed innovation. They have described how they used digital chemistry in their efforts to design more sustainable materials and how this approach has sped up timelines by 10x on average compared to a solely experimental approach.
Physics-Based Simulations to Develop New Energy Solutions
Another exciting application of computational chemistry approaches is the use of atomic-scale materials modeling in the design of new battery and energy storage solutions.
Some behaviors of materials that have been studied include ion diffusion, electrochemical response in electrodes and electrolytes, dielectric properties, mechanical response, and more. This computational approach has been used to screen for Li-ion battery additives that form a stable solid electrolyte interphase.
Driving R&D with Schrödinger’s Pioneering Computational Platform
At Schrödinger, our physics-based computational platform allows companies worldwide to harness the capabilities of computational chemistry methods and apply these to their R&D programs quickly and with ease. Over the last 30 years, Schrödinger’s modeling software and services have enabled the discovery of high-quality, novel molecules and materials across industries–as illustrated by some of the examples described above.
Molecules come to life in Maestro, the streamlined portal for structural visualization and access to cutting-edge predictive computational modeling and machine learning workflows. And researchers can bring their digital and experimental data side-by-side within LiveDesign, Schrödinger’s enterprise informatics platform for collaborative analysis, molecular design, and program management.
As the predictive and analytical capabilities of physics-based modeling continue to advance and are enhanced by the addition of new ML models, the myriad applications that are impacted by computational chemistry will continue to grow.
References
-
Advancing Drug Discovery through Enhanced Free Energy Calculations
2017. Abel R, Wang R, Harder ED, Berne BJ, and Friesner RA. Accounts of Chemical Research. 50(7):1625-1632. DOI: 10.1021/acs.accounts.7b00083
-
Docking and scoring in virtual screening for drug discovery: methods and applications
2004. Kitchen D, Decornez H, Furr J, et al. Nature Review Drug Discovery. 3:935–949. DOI: 10.1038/nrd1549
-
Efficient Exploration of Chemical Space with Docking and Deep Learning
2021. Yang Y, Yao K, Repasky MP, Leswing K, Abel R, Shoichet BK, and Jerome SV. Journal of Chemical Theory and Computation I. 17(11): 7106-7119. DOI: 10.1021/acs.jctc.1c00810