Exploring the effects of wetting and free fatty acid deposition on an atomistic hair fiber surface model incorporating Keratin Associated Protein 5-1
Shearing Friction Behaviour of Synthetic Polymers Compared to a Functionalized Polysaccharide on Biomimetic Surfaces: Models for the Prediction of Performance of Eco-designed Formulations
DeepautoQSAR hardware benchmark

DeepAutoQSAR hardware benchmark

Executive Summary
- This benchmark evaluates the performance of DeepAutoQSAR on two datasets of different sizes using different hardware configurations and model training times.
- Our general recommendations, based on the results and the hardware costs, are to use the NVIDIA T4 GPU hardware with the following training times: 2 hrs for datasets with less than 1,000 data points; 4 hrs for 1,000 to 10,000 data points; and 8 hrs for more than 10,000 data points.
- While performance ultimately depends on the data, the intended purpose of this benchmark is to serve as a starting point for choosing the hardware to train the ML model(s) with and the specific model training time to use. Actual performance is highly dependent on the specific dataset and may require increasing the training time or choosing a different GPU to achieve the desired results.
Introduction
The application of machine learning (ML) to predict the molecular properties of drug candidates is an important area of research that has the potential to reduce drug development timelines and accelerate the creation of medicines for patients with serious unmet medical needs.
The successful application of ML relies on sufficient data quantity and quality, a suitable model architecture(s) for the given problem, proper hyperparameter choices (the parameters for a particular ML model architecture), and appropriate model training time for a chosen hardware configuration.
DeepAutoQSAR is a machine learning product that allows users to predict molecular properties based on chemical structure. The automated supervised learning pipeline enables both novice and experienced users to create and deploy best-in-class quantitative structure activity/property relationship (QSAR/QSPR) models.
The purpose of this benchmark, which builds on the work of an earlier whitepaper [1], is to characterize the performance of DeepAutoQSAR on two datasets of different sizes using different hardware configurations and model training times. While performance ultimately depends on the data, the intended purpose of this benchmark is to serve as a starting point for choosing the hardware to train the ML model(s) with and the specific model training time to use.
Datasets
The datasets used in the benchmark were obtained from the Therapeutics Data Commons (TDC). TDC provides ML-ready datasets that can be used for learning tasks that are valuable to pharmaceutical research and development and that cover different therapeutic modalities and stages of the drug development lifecycle [2].
We use two datasets that contain assay data for one Absorption, Distribution, Metabolism, and Excretion (ADME) property each:
- Caco2 (Human Epithelial Cell Effective Permeability)
- AqSolDB (Aqueous Solubility)
Performance is measured by the median accuracy of the ADME property prediction for a sample of train-test data splits; note that the specific train-test data splits used are different from the splits provided by TDC for its benchmark leaderboard.
Dataset Descriptions
Caco2 (Human Epithelial Cell Effective Permeability) [3]*
The human colon epithelial cancer cell line, Caco-2, is used as an in vitro model to simulate the human intestinal tissue. The experimental result on the rate of drug passing through the Caco-2 cells can approximate the rate at which the drug permeates through the human intestinal tissue.
This dataset contains numerical data for use in regression, and there are 906 compounds.
AqSolDB (Aqueous Solubility) [4]*
Aqueous solubility measures a drug’s ability to dissolve in water. Poor water solubility could lead to slow drug absorptions, inadequate bioavailability and even induce toxicity. More than 40% of new chemical entities are not soluble.
This dataset contains numeric, non-integer data for use in regression, and there are 9845 compounds.
*Note: The datasets have been modified from their original form to remove structural redundancies and experimental errors.
Hardware
The hardware used in the benchmark was provisioned from the Google Cloud Platform (GCP); therefore, the hardware configurations chosen were based on the machine types offered by Google.
These limitations on hardware configurations, dictated by the cloud provider, mean that only specific hardware pairings are available, such as a particular GPU platform that can only be used with a given CPU platform. For example, NVIDIA A100 GPUs can only be run on an A2 machine type, which only uses the Intel Cascade Lake CPU platform. Constrained by these limitations, every effort was made to keep hardware-specific options consistent across machine types, to provide hardware diversity when reasonable, and to use cost-effective high-performance computing hardware.
| Hardware Key | GCP Machine Type | CPU Platform | vCPUs* | RAM (GB) | GPU Platform | GPUs | Cost ($) per Hour+ |
|---|---|---|---|---|---|---|---|
| 2 vCPUs | n2-standard-2 | Intel Ice Lake | 2 | 8 | N/A | None | $0.10 |
| 4 vCPUs | n2-standard-4 | 4 | 16 | $0.19 | |||
| 8 vCPUs | n2-standard-8 | 8 | 32 | $0.39 | |||
| 16 vCPUs | n2-standard-16 | 16 | 64 | $0.78 | |||
| T4 GPU | n1-standard-4 | Intel Ice Lake** | 4 | 15 | Nvidia T4 | 1 | $0.54 |
| V100 GPU | Nvidia V100 | $2.67 | |||||
| A100 GPU | a2-highgpu-1g | Intel Cascade Lake | 12 | 85 | Nvidia A100 | $3.67 |
** Up to Intel Ice Lake generation; GCP auto assigns CPU platform on node pool creation.
+ Prices in November 2022. Includes sustained use discounts.
Benchmarking Methods & Results
Our benchmark is a two stage process. In the first stage, DeepAutoQSAR models are trained to fit the TDC datasets using a standard cross validation procedure to select top performing ML models for the model ensemble and to optimize hyperparameters; the end result of this stage is an ensemble of top performing models, which, under normal usage, are averaged to provide a mean prediction and associated ensemble standard deviation. We detail the specific protocol in our white paper, a Benchmark Study of DeepAutoQSAR, ChemProp, and DeepPurprose on the ADMET Subset of the Therapeutic Data Commons [1]. In the second stage, random train-test splits of the data are computed, and the previously determined ensemble of top ML models architectures with specific hyperparameter configurations are trained on the new training data splits. Predictions are then generated for the new test data splits. These multi-split metrics provide a more robust estimate of model performance by reducing potential bias introduced from a single train-test data split. Model performance in this hardware benchmark is reported as the median R2 coefficient of determination [5] across these random train-test splits for each hardware configuration and model training time.
In the first stage, the initial training procedure runs continuously for each training time allotment. Due to the stochastic nature of hyperparameter optimization and model architecture selection, each hardware and training time combination can potentially explore a different number of model architectures and hyperparameter combinations each time a benchmark job is run. The model training times evaluated were: 0.5, 1, 2, 4, 8, and 16 hours. As a general rule, more competent hardware running for longer training times on smaller datasets (e.g., a machine with an A100 GPU training for 16 hrs on the smaller Caco2 permeability dataset) will explore more hyperparameterizations than less competent hardware running for shorter training times on larger datasets (e.g., a two core machine training for 2 hrs on the larger AqSolDB dataset).
Since model architecture selection and hyperparameter sampling is a stochastic process, we run each benchmark configuration, which is the particular hardware and training time combination, three times and report averages for performance—this is especially relevant when fewer hyperparameter combinations are explored as model performance is more sensitive to hyperparameter sampling. The output of the first stage is an ensemble of top models, determined by cross validation, with specific hyperparameters choices for each.
The second stage of our benchmark runs for half the training time of the first stage. Increasing training time leads to more robust statistics as the median performance converges to a split-independent value, but comes at the expense of increased computational cost; in practice computational expense must be balanced with the need to train the ensemble model for a sufficiently large training time. For performance reporting we provide the median R2 coefficient of determination [5] as computed from the multiple train-test splits, which aims to reduce potential bias introduced by a single train-test split. To compute this R2, we repeatedly split the data into training and testing sets via bootstrap sampling with replacement; to do so, we take N samples with replacement from the dataset with N total data points and remove any duplicates to form a subset. The selected points are then used to train the specific model architectures found in stage one, and the unselected points serve as the test holdout. We do this until the time limit is reached and report the median R2 of all resamplings.
As both of the TDC datasets are numerical regression problems, this metric is a reasonable measure of model performance; however, the choice of performance metric in real-world applications should always be determined according to the use-case of the ensemble model. Sometimes MAE or RMSE are more appropriate to assess if a model is sufficiently performant. The output of the second stage is a distribution of ensemble model performances over different train-test splits; the reported value is the median of the distribution.
We plot the benchmark results, which is the median R2 coefficient of determination from the second stage, below. Our first plot shows performance on the AqSolDB dataset, and the second plot shows performance on the Caco2 permeability dataset. For each of these datasets, we highlight the progression of performance over time grouped by hardware type, where hardware type is on the x-axis, training time in hours is the bar color, and median R2 score is on the y-axis. The data used to generate the plots are provided in the supplementary tables.
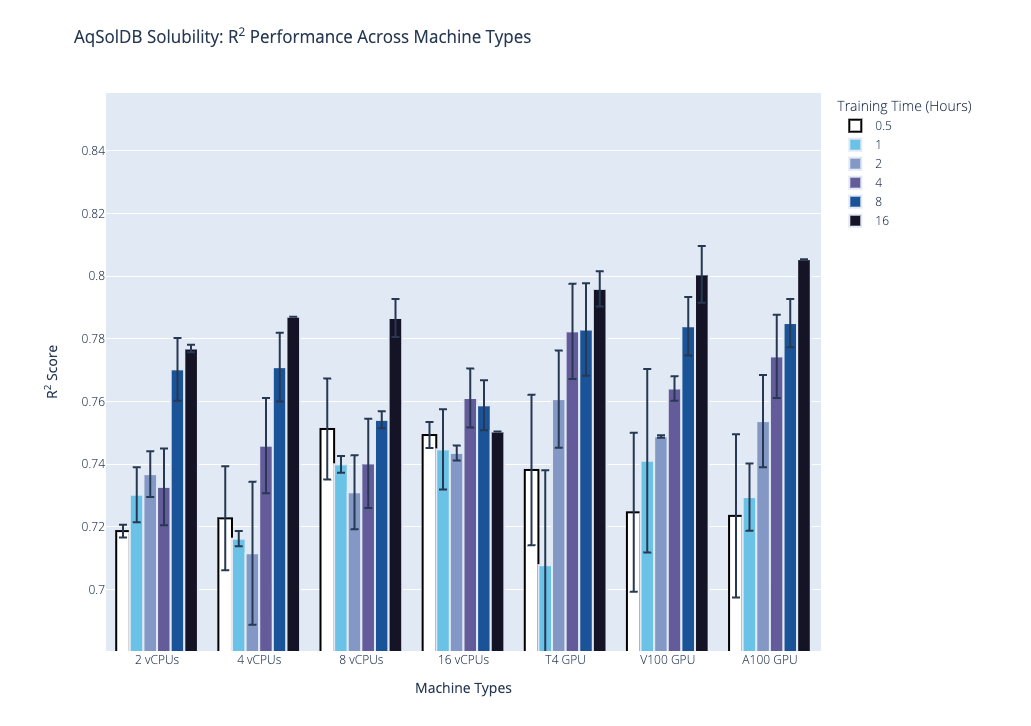
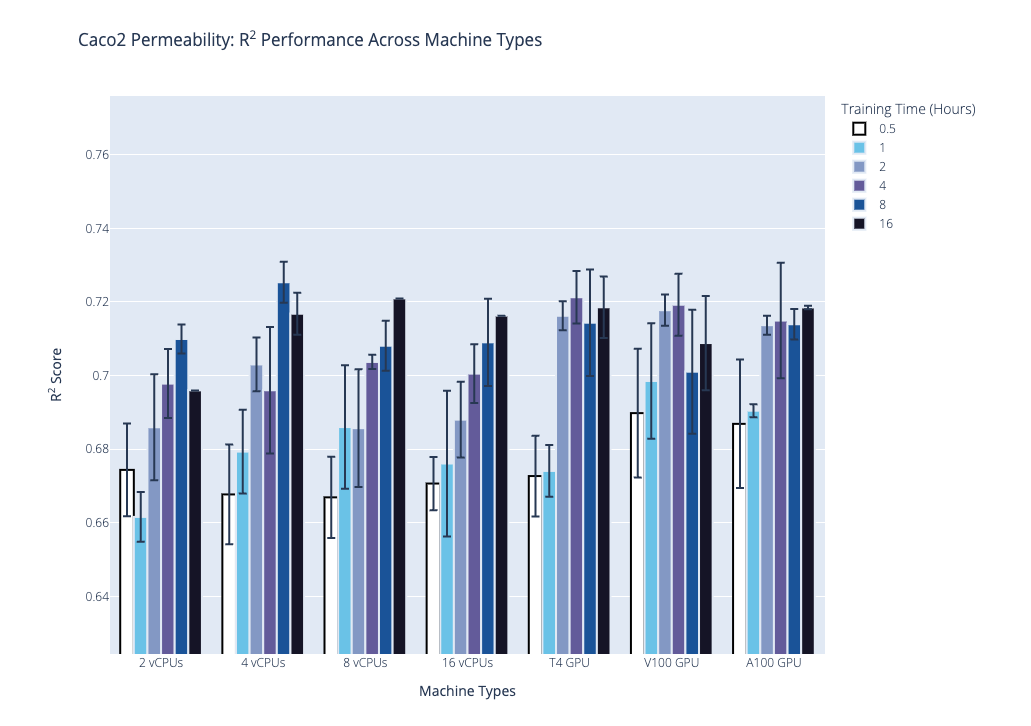
Based on these results and the hardware costs, our general recommendations are the following:
| Number of Data Points | Hardware | Training Time (hr) |
|---|---|---|
| <1,000 | Nvidia T4 GPU | 2 |
| 1,000 – 10,000 | 4 | |
| >10,000 | 8 |
Selected publications
-
Kaplan, Z.; Ehrlich, S.; Leswing, K. Benchmark study of DeepAutoQSAR, ChemProp, and DeepPurpose on the ADMET subset of the Therapeutic Data Commons. Schrödinger, Inc., 2022.
https://www.schrodinger.com/science-articles/benchmark-study-deepautoqsar-chemprop-and-deeppurpose-admet-subset-therapeutic-data (accessed 2022-11-29).
-
Therapeutics Data Commons.
https://tdcommons.ai/ (accessed 2022-06-15).
-
ADME – TDC.
https://tdcommons.ai/single_pred_tasks/adme/#caco-2-cell-effective-permeability-wang-et-al (accessed 2022-06-15).
-
ADME – TDC.
https://tdcommons.ai/single_pred_tasks/adme/#solubility-aqsoldb (accessed 2022-06-15).
-
Sklearn.metrics.r2_score — scikit-learn 1.1.3 documentation.
https://scikit-learn.org/stable/modules/generated/sklearn.metrics.r2_score.html#sklearn-metrics-r2-score (accessed 2022-11-29).
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
The role of digital chemistry across the polymer supply chain

The role of digital chemistry across the polymer supply chain
Molecular modeling and simulation tools have proven effective in materials development and are increasing in use throughout the polymer industry, from raw materials suppliers to end product manufacturers. Computational workflows also open new avenues for developing polymers with improved recyclability. Physics-based simulations offer reliable predictions of structures, morphologies, properties, and chemical reactivity for polymers. Recent advances in machine learning, deep learning, and enterprise informatics platforms have accelerated the speed, accuracy, and automation of novel materials and solutions discovery. A paradigm shift to computer-driven molecular design is occurring throughout the industry.

Vision of a digital chemistry catalyzed polymer supply chain:
Raw Materials Suppliers: Employ atomistic simulations to improve understanding and predict properties of downstream products; offer optimized raw materials to compounders.
Polymer Compounders: Use atomistic simulations to understand formulation chemistry and predict properties; provide detailed requirements to raw materials suppliers and offer optimized products to end product manufacturers.
End Product Manufacturers: Leverage atomistic simulations to predict the performance of final products and quickly identify causes of failure; give specific formulation requirements to compounders.

Solution Overview
Schrödinger’s Materials Science platform offers tailored solutions for research and business throughout the polymer supply chain, with differentiated model builders, efficient simulation engines accelerated by GPU computing power, automated thermophysical and mechanical response workflows, and accurate analysis tools.
- Broad molecular simulation and property prediction tools for: Thermal Properties, Mechanical and Dielectric Properties, Reactivity and Kinetics, Aggregation in Polymer Production, Solvent Sensitivity, Gas, Ion, Additive Diffusivity, Phase Morphology and SAXS Scattering, Semi-crystalline Morphology
- Applicable to all polymer types: thermoplastic homo and copolymers, crosslinked, elastomers, and dendrimers
- Intuitive user interface with automated workflows for experts or non-experts
- Dedicated scientific/technical support and vast learning resources
Digital Chemistry Value Across Polymer Supply Chain (Example: Transportation Industry)
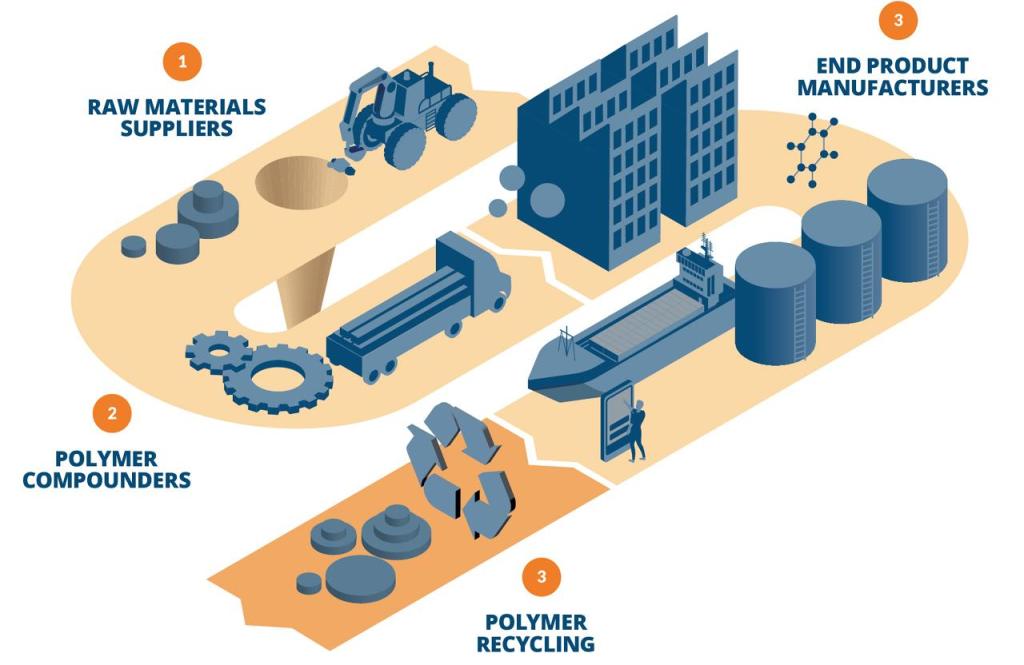
1. Raw Materials Suppliers
Suppliers of petrochemical and chemical feedstocks, additives, and various monomers and resins
Design new chemistries from alternative sources and discover new applications through simulating downstream products properties
- Predict polymer crosslinker performance in composite matrix resins such as epoxy-amine and cyanate esters
- Simulate the interaction between thermoplastic styrene-butadiene and crosslinkers
Speed decision making for catalyst selection in raw materials production
- Simulate and understand the catalysis mechanisms, selectivity, and reactivity of epoxy amine, urethane, and other reactions
Develop alternative greener raw materials that are more environmentally sustainable
- Simulate the impact of degradation on modulus for a chemistry of focus
2. Polymer Compounders
Suppliers who prepare polymer formulations by mixing or/and blending polymers and additives into process-ready products
Predict the performance of alternative raw materials in formulations and end products
- Predict glass transition, thermal stability, and thermal expansion with new polymers
- Quantify the diffusion of additive in polymers • Understand water transport and morphological stability of polymer formulations
Efficiently optimize formulation properties
- Predict and track water uptake in polymer composites
- Predict curing kinetics and processing properties
Develop greener formulations that are more environmentally sustainable
- Simulate and screen for optimal formulation with new bio-based chemistry
3. End Product Manufacturers
Processors of resins/formulations who make them into finished products on the market
Enable reliable decision-making through predictive modeling of end product properties
- Predict tire materials performance with different additives and cross-linkers
Obtain best chemistry from upstream suppliers by targeted chemical design to properties critical to product and processing constraints
- High-throughput screening of epoxy-amine reactions to identify the unique combinations for target properties
Accelerate the manufacturing process pipeline
- Predict polymer gelling during manufacturing process
Quickly screen and identify potential causes and impacts of manufacturing and material source deviations
- Predict sensitivity of matrix to cleaning solvents
Design greener products that are more environmentally sustainable
- Simulate and predict properties of high-performance resins with bio-based materials and automate discovery of new biomaterials
4. Polymer Recycling
Research and design for recyclability throughout the polymer supply chain
Design polymers for recyclability
- Predict selectivity of chemical recycling reaction
Expand use of recycled materials
- Simulate impact of recycled polymers in packaging
Determine impact on product with use of recycled material
- Screen for property changes with recycling driven microstructure changes
About Schrödinger
Schrödinger is transforming the way materials are discovered. Schrödinger has pioneered a physics-based software platform that enables discovery of high-quality, novel molecules for materials applications more rapidly and at lower cost compared to traditional methods.
Learn how digital chemistry is driving innovation across materials science industries
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Innovation in atomic-level processing with atomistic simulation and machine learning
Innovation in atomic-level processing with atomistic simulation and machine learning
Background
The Schrödinger Materials Science Suite offers computational tools specifically tailored to studying the gas-surface chemistries of atomic layer deposition and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (usually with density functional theory, DFT) and prediction of key properties. This modeling approach thus has a unique role to play both in deepening our understanding of existing processes and in discovering novel chemicals.
Many of today’s high-tech devices are manufactured by processing materials at the nanoscale, including computer chips, data storage and communications devices, sensors, solar cells and batteries. Making devices smaller, more powerful and more energy-efficient means developing new patterning, deposition and etch techniques at ever-finer resolution, in some cases down to just a few atoms thick. Chemical processing is being pushed to its limits in terms of purity, uniformity, conformality and substrate-selectivity.
Optimizing a deposition or etch process to more stringent tolerances requires a deep understanding of the underlying chemistry. Integrating new materials into a device often requires the discovery of new chemicals with improved properties. In both respects, researchers are increasingly turning to computer simulations to cut down on lab work and make discoveries faster.
The Schrödinger Materials Science Platform offers computational tools specifically tailored to tackling this problem, by studying the gas-surface chemistries of deposition, etch and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (with density functional theory, DFT) and prediction of key properties. This modeling approach can both deepen our understanding of existing processes and help discover novel chemicals.
Automate Precursor Screening for Improved Properties
Organometallic complexes can be used to deposit or etch pure metals or compounds such as oxides or nitrides. However, synthesis of the complexes in the lab can be time-consuming and difficult because many complexes react violently in air. Subsequent characterization and testing in a reactor is even more costly. Digital chemistry can rapidly screen larger numbers of chemicals than can ever be experimentally tested, narrowing down the chemical space and discovering the most promising candidate molecules for deposition or etch.
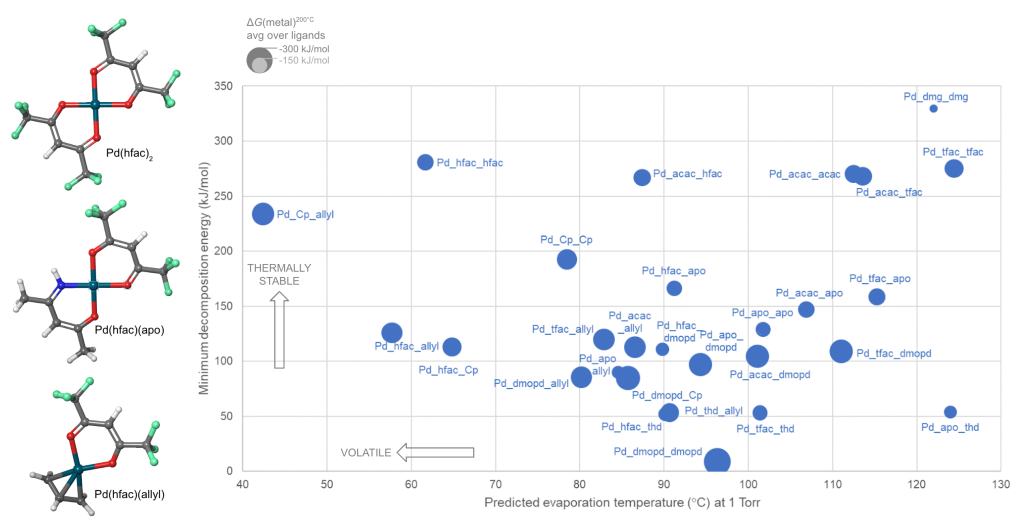
Schrödinger provides easy-to-use software for automated generation and assessment of hundreds or thousands of precursor gases. A library of hundreds of commonly-used ligands is provided, including bidentate and haptic ligands, and the library can be customized. A selection from the library is then used to enumerate and build all possible precursor complexes. We present here a case study in which a library of 10 ligands form 55 distinct complexes with divalent palladium (Pd) as the metal cation and some of these candidate complexes are shown in Figure 1.
Synthesizability
Combining two or more different ligands in a heteroleptic complex is a promising way to tune precursor properties, but it is difficult to guess whether a given combination can in fact be synthesized. This question can be answered by computing the energy for ligand exchange back to homoleptic complexes. In the Pd example, this reveals that 14 of the heteroleptic complexes are unstable and can not be synthesized.
Reactivity
Precursors for deposition must be carefully designed so as to react cleanly and deposit the desired solid material, via thermolysis or plasma activation (chemical vapor deposition, CVD) or via reactions with adsorbed fragments (atomic layer deposition, ALD). Likewise, reagents for etching must react with surface atoms to form gaseous products. Lists of candidate precursor molecules can be efficiently screened against deposition or etch chemistries using Jaguar, Schrödinger’s code for molecular DFT. Figure 1 displays the computed reactivity of the complexes with respect to Pd deposition.
Volatility and Thermal Stability
To be useful in a real CVD or ALD process, a precursor must also be volatile and thermally stable during storage and delivery. This limits the process to a window of viable temperatures, with insufficient vapor pressure at lower temperatures and impurities from decomposition at higher temperatures. For instance, Pd(thd)2 suffers from decomposition when heated to 200-250°C for the ALD of Pd metal.1, 2 Calculating volatility and thermal stability with Schrödinger’s tools gives information on how to tune these properties and widen the temperature window for maximum process flexibility.
Schrödinger has used machine learning (ML) to develop a unique model for predicting the evaporation or sublimation temperature of organometallic complexes at a given pressure. The ML-volatility model is accurate to an average of ±9°C over the training set and can evaluate hundreds of molecules per second.
The thermal stability of precursor gases can be quantified by considering the unimolecular decomposition channels: bond dissociation and β-H elimination. All such reactions in each molecule are automatically computed at the DFT level and the lowest energy reaction is selected as the most likely. The minimum decomposition energy of each complex is thus a measure of its thermal stability.
These approaches were applied to compute the properties of the Pd complexes, and Figure 1 shows how this allows the candidate precursors to be assessed.
Rational innovation is facilitated by Schrödinger’s atomistic modeling tools. Novelty is maximized by using a large library of ligands to build a wide variety of complexes, homoleptic and heteroleptic. Risk is minimized by selecting for lab study only those precursors computed to properly combine the desired properties of synthesizability, volatility, reactivity and stability.
Gain Insights Into Competing Processes for Optimum Deposition Conditions
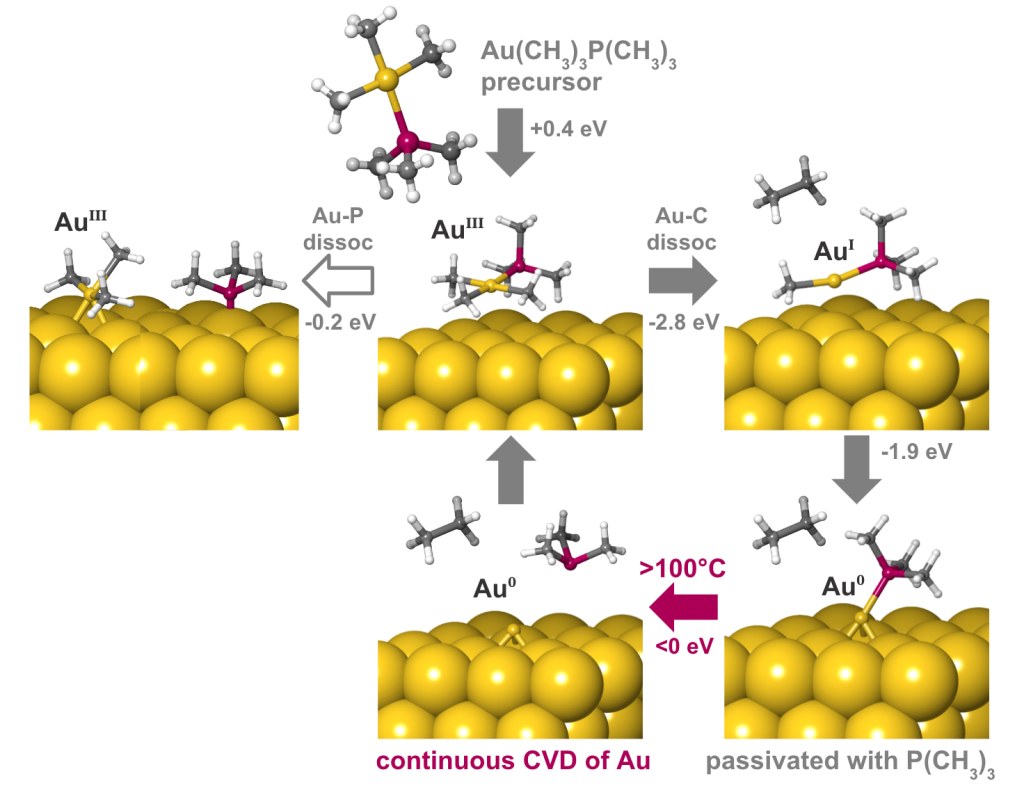
A deposition or etch process consists of a sequence of individual reaction steps, often in competition with other reactions. Simulations give atomic-level insight into these complicated mechanisms and how they depend on reactor conditions such as temperature and pressure. Here we investigate the mechanism of gold deposition from the single-source precursor trimethylphosphinotrimethylgold(III). Experiments show that gold films grow in a self-limiting fashion when this precursor is pulsed at low temperature, switching over to continuous CVD at 100°C and higher, but the reason is unknown.3
To explain this behavior, the Quantum ESPRESSO code for periodic DFT has been used to examine the detailed mechanism of reactive adsorption of the precursor onto a metallic gold surface. Thermodynamics of competing reaction steps have been computed as a function of process temperature and pressure with Schrödinger’s tools for adsorption and surface chemistry [4].
The results are summarized in Figure 2. The physisorbed precursor is in a metastable state, from which dissociation of Au-P or Au-C bonds can lead to chemisorption. Au-C dissociation and elimination of C2H6 is found to be the favored pathway, due to a strong thermodynamic driving force towards reduction of Au(III) to Au(I) and eventually Au(0). Trimethylphosphine is computed to bind to the surface, forming an adlayer that passivates against further precursor adsorption. This explains the low rate of self-limited deposition at <100°C. The computed free energies also reveal that desorption of trimethylphosphine becomes favored at higher temperatures, in agreement with the crossover to CVD seen experimentally at >100°C.
Finding out which bond breaks and how the surface is passivated opens up chemically-informed strategies for improving the process, for example by choosing a suitable co-reagent or modifying the precursor structure. Digital chemistry allows rational design to take the place of trial-and-error.
This example illustrates how changes in process behavior can be traced back to competing chemical pathways. DFT simulations give unique qualitative insight into atomic structure, bonding and oxidation states of the intermediates along each reaction pathway. In addition, quantitative information about kinetics and thermodynamics as a function of process temperature can be computed, giving valuable guidance on how to optimize the nanofabrication process. The result is improved control of purity, uniformity, phase and substrate-selectivity.
Conclusion
These cases illustrate how Schrödinger’s Materials Science Platform provides powerful and customizable computational solutions to accurately model surface reactivity, predict precursor properties and discover novel chemicals for optimized materials deposition or etch processes.
References
-
Low temperature atomic layer deposition of noble metals using ozone and molecular hydrogen as reactants,
J. Hämäläinen et al., Thin Solid Films 2013, 531, 243-250, DOI 10.1016/j.tsf.2013.01.091.
-
Atomic layer deposition of noble metals: Exploration of the low limit of the deposition temperature,
T. Aaltonen et al., J. Mater. Res. 2004, 19, 3353–3358, DOI 10.1557/JMR.2004.0426.
-
Reaction mechanism of the Me3AuPMe3–H2 plasma-enhanced ALD process, M. Van Daele et al.,
Phys. Chem. Chem. Phys. 2020, 22, 11903-11914, DOI 10.1039/C9CP06855D.
-
First-principles investigation of crossover between ALD and CVD in the thin film deposition of gold,
C. N. Brock. D. J. Giesen, A. Fonari & S. D. Elliott, Materials Research Society Spring Meeting, 2022.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Are Deep Learning Structural Models Sufficiently Accurate for Free-Energy Calculations? Application of FEP+ to AlphaFold2-Predicted Structures
Pathfinder-Driven Chemical Space Exploration and Multiparameter Optimization in Tandem with Glide/IFD and QSAR-Based Active Learning Approach to Prioritize Design Ideas for FEP+ Calculations of SARS-CoV-2 PLpro Inhibitors
Overview of Molecular Modelling for Formulations

DEC 19, 2022
Overview of Molecular Modelling for Formulations
Speaker
John Shelley
Fellow
Summary
This talk is an overview of molecular modeling calculations relevant for formulations in the pharmaceuticals, inks, 3D printing, polymers, batteries and agricultural chemicals industries.
Chinese: 2022薛定谔秋季中文生命科学网络讲座 | 基于物理理论的计算模拟 – 如何准确预测小分子晶体的结构和溶解度


DEC 7, 2022
2022薛定谔秋季中文生命科学网络讲座 | 基于物理理论的计算模拟 – 如何准确预测小分子晶体的结构和溶解度
Speakers
Lingle Wang
Vice President
Abstract
对固态科学家来说,药物晶体形式的改变是药物研发后期甚至上市后是非常严重的,打击性极强的问题。这类新型的晶体形式可能表现出不同的,有可能是负面的特性。许多药物的多晶型消失,包括利托那韦、罗替戈汀和盐酸雷尼替丁等,给制药公司带来了巨大的损失并引发一系列患者诉讼。本次研讨会,我将介绍一种通过大规模回顾性验证和被实际药物制剂过程中的前瞻性研究所证明的、可靠且准确的方法来预测候选药物的所有低能量稳定多晶型物及其相对稳定性。此外,我们还将重点介绍通过自由能扰动 (FEP+) 方法准确计算晶体结构的热力学溶解度。
Emergence of unexpected crystal forms during late stages of drug development or after the initial launch of the drug is a very frustrating and serious problem for solid-state scientists. Since the newly emerged forms can exhibit different and potentially undesired properties, disappearing polymorphs for many drugs, including ritonavir, rotigotine and ranitidine hydrochloride for example, have caused big losses for pharmaceutical companies and series patient litigations. In this presentation, I will present a reliable and accurate method to predict all the low energy stable polymorphs of a given drug candidate and their relative stabilities, as demonstrated both from large scale retrospective validation and prospective studies in real drug formulation processes. I will also highlight the accurate calculations of the thermodynamic solubilities of crystal structures from free energy perturbation (FEP+) method.
Chinese: 2022薛定谔秋季中文生命科学网络讲座 | 用最新的基于物理计算方法为基于结构的药物研发开辟新天地

NOV 24, 2022
2022薛定谔秋季中文生命科学网络讲座 | 用最新的基于物理计算方法为基于结构的药物研发开辟新天地
Speaker
Dr. Jianxin Duan
Fellow
Abstract
近年来,随着新的高预测性、基于物理理论方法的发展与其加速发现新型临床化合物能力的展现,基于结构的药物发现 (SBDD) 策略的价值得到提升。然而,这些方法受靶蛋白的高质量结构模型可用性的限制。 最新的结构生物学创新利器,如冷冻电镜和计算预测的蛋白质模型(使用机器学习和基于物理的方法)有望开创一个新的靶点纪元。 在本次网络研讨会中,我们将介绍最新的计算工作流程如何在这些具有历史挑战性的靶点和脱靶点上实现基于结构的药物发现。
主要议题:
在没有实验晶体结构(即同源模型或 AlphaFold 结构)的情况下,建立和验证高质量蛋白质结构模型用于SBDD的新计算方法。
通过以下案例展示新方法在项目中的作用:
1)推进用高通量筛选获得的初步苗头化合物
2)解决脱靶效应带来的障碍
3)使用同源模型推进整个项目
The value of pursuing a structure-based drug discovery (SBDD) strategy has amplified in recent years as new highly-predictive, physics-based methods have evolved and demonstrated the ability to accelerate the discovery of novel clinical compounds. However, these approaches are limited by the availability of high-quality structural models of the target protein. Recent advances in structural biology such as cryo-EM and computationally-predicted protein models (using machine learning and physics-based methods) have the potential to open a new world of targets to pursue. In this webinar, you’ll learn how new advances in computational workflows are enabling structure-based drug discovery on these historically challenging targets and off-targets.
Key topics covered:
Overview of new computational approaches for building and validating high-quality protein structural models for use in SBDD in the absence of an experimental crystal structure (i.e. homology models or AlphaFold structures)
Case studies demonstrating the impact of these approaches to:
1) progress initial hits from high-throughput screens
2) dial-out off target liabilities
3) progress entire programs using homology models
Driving innovation in polymer R&D with molecular simulation & machine learning
Driving innovation in polymer R&D with molecular simulation & machine learning
R&D scientists across broad industries from aerospace to electronics face challenges in developing the next-generation of polymers and composites with high-performance, multifunctional capabilities that also meet society’s demands for miniaturization and environmental sustainability.

Solution Overview
Chemical reactivity, physical morphology, and polymer physics drive the behavior of polymers and soft materials. Schrödinger’s Materials Science platform provides a unique combination of capabilities for the design and optimization of polymers, including:
- Differentiated model builders
- Extremely efficient, GPU-powered molecular dynamics (MD) engine
- Automated thermophysical and mechanical response workflows
- Chemically adaptable cross-linking workflows
- Analysis tools for the simulation, optimization, and discovery of novel polymers including linear, crosslinked, elastomers, dendrimers, and copolymers
Schrödinger’s tailored solutions for molecular modeling of polymers, mixtures, and composites can reduce cost and risk, shorten timelines and drive innovation in a broad range of industries.
Industries and Applications
Automotive and Aerospace
- Optimize polymer formulation properties: Model water transport, uptake and morphological stability in polymer composites using multistate simulations
- Accelerate chemical design to properties critical to product and processing constraints: Identify the unique combinations of epoxy-amine reactions for target properties using highthroughput screening
- Accelerate the manufacturing process pipeline: Predict polymer gelling during manufacturing process
- Design greener products that are more environmentally sustainable: Simulate and predict properties of high-performance resins with bio-based materials, and automate discovery of new biomaterials
CPG Packaging and Formulation
- Develop environmentally sustainable packaging: Study interfacial interactions between packaging materials and consumer goods, and simulate water uptake for barrier design and performance
- Optimize polymer formulation: Quantify the diffusion of additives in polymer packaging materials
- Minimize production waste: Understand the potential causes and impact of polymer production deviations
- Innovate with natural materials: Explore active, recyclable, and bio-based materials through molecular simulation
- Reduce processing cost: Evaluate new formulations for potential manufacturing challenges
Specialty Polymers
- Speed up decision-making on catalyst selection for raw materials production: Simulate and understand the catalysis mechanisms, selectivity, and reactivity of epoxy amine, urethane, and other reactions
- Optimize the design of high-performance polymers: Predict glass transition, thermal stability, and thermal expansion with new polymers
- Drive innovation in polymer reactions: Predict curing kinetics and processing properties
Electronic Packaging
- Identify thermal/mechanical performance issues early in design: Simulate coefficient of thermal expansion for packaging polymers
- Optimize chemical stability: Simulate interactions of packaging polymers with processing solvents and water to predict stability in use
- Design new monomers for improved performance: Predict design drivers like Dk, Df and refractive index with physics-based simulations and machine-learned models
Batteries
- Understand mechanisms at molecular level: Predict electrolyte degradation reaction mechanisms and control energetics to understand the formation of functional solid electrolyte interphase layers at anodes (SEI) and cathodes (CEI)
- Identify best-performing battery materials: Perform high-throughput screening of anode, cathode, electrolyte and additive materials to identify best chemistries for development of nextgeneration batteries
- Optimize electrolyte design: Simulate interatomic interactions and transport of ions in liquid and polymer electrolytes
Pharmaceutical Formulation
- Optimize drug carrier design: Evaluate the solubility/miscibility of drugs with different polymeric carriers or barriers
- Optimize drug formulations: Visualize and characterize pH-dependent polymeric surfactant and drug interactions in solution and precipitation inhibition behavior
- Drive effective drug delivery: Simulate drug release profiles from amorphous solid dispersions into solution
Team Collaboration and Digital Data Management
- Empower collaboration: Employ web-based enterprise informatics tools for sharing experimental and predictive models seamlessly
- Amplify research: Rapidly deploy of machine learning models to drive predictions and assist novel design approaches
- Improve project management: Accelerate project communication and collective learning by capturing, analyzing, and testing new ideas and data in a centralized platform
Products
Check on the products that enable your success in the polymer industry.
References
-
Melt-state degradation mechanism of poly (ether ketone ketone): the role of branching on crystallization and rheological behavior.
Wiggins J. et al. Polym. Degrad. Stab. 2022, 200, 109968.
-
Atomistic Molecular Dynamics in PEO/PMMA Blends Having Significantly Different Glass Transition Temperatures.
Habasaki J, Int. J. Appl. Glass Sci, doi.org/10.1111/ijag.16553.
-
Experimental Investigations of AlMg3 Components with Polyurethane and Graphene Oxide Nanosheets Composite Coatings, after Accelerated UV-Aging.
Murariu A. C. et al. Molecules 2022, 27, 1, 84.
-
Polycyanurates via Molecular Dynamics: In Situ Crosslinking from Di(Cyanate Ester) Resins and Model Validation through Comparison to Experiment.
Moore L. M. J. et al. Macromolecules 2021, 54, 13, 6275–6284.
-
Photophysical Properties of Cyclometalated Platinum(II) Diphosphine Compounds in the Solid State and in PMMA Films.
Anderson C. M. et al. ACS Omega 2021, 6, 42, 28316–28325 High-Throughput Molecular Dynamics.
-
Simulations and Validation of Thermophysical Properties of Polymers for Various Applications.
Afzal. A et al. ACS Appl. Polym. Mater. 2021, 3, 2, 620–630 Fundamental Limits to the Electrochemical Impedance Stability of Dielectric.
-
Elastomers in Bioelectronics.
Floch P. L. et al. Nano Lett. 2020, 20, 1, 224–233.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.