AutoTS
Automatic workflow for locating transition states for elementary reactions

Automatic workflow for locating transition states for elementary reactions
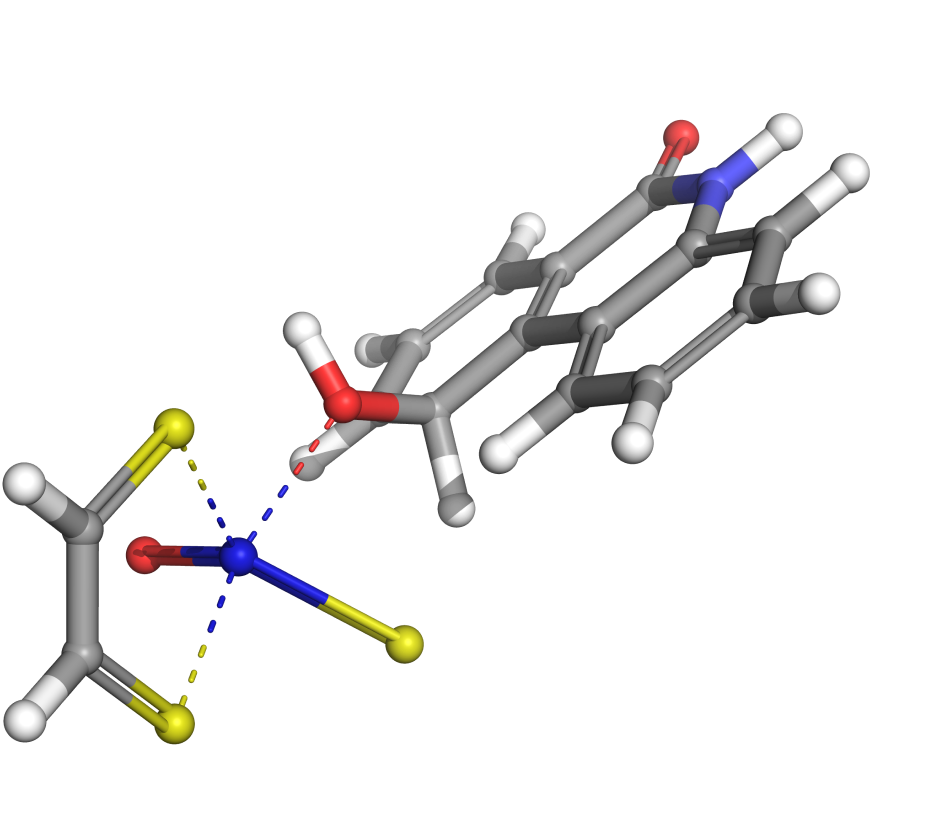
Transition states are essential in many materials science applications: predicting reactivity, understanding reaction mechanisms, designing and optimizing catalysts, predicting outcomes of various competing reactions, and more. Locating a transition state (TS) is necessary for computing the activation energy of a reaction, and thereby the reaction rate, and it is unique to computation – meaning that the transition state cannot be “found” in the lab.
AutoTS is an automated workflow to find transition states, particularly for elementary, molecular reactions. AutoTS requires only the structures of the reactants and the products as input, and then automates the search process to obtain the transition state and the reaction energetics.

Get more from your ideas by harnessing the power of large-scale chemical exploration
and accurate in silico molecular prediction.
Get answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
Quantum mechanics solution for rapid and accurate prediction of molecular structures and properties
Automated workflows for design, optimization, and unsupervised mechanism discovery in molecular chemistry
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.