Shape Screening
Efficient ligand-based virtual screening of millions to billions of molecules

Efficient ligand-based virtual screening of millions to billions of molecules
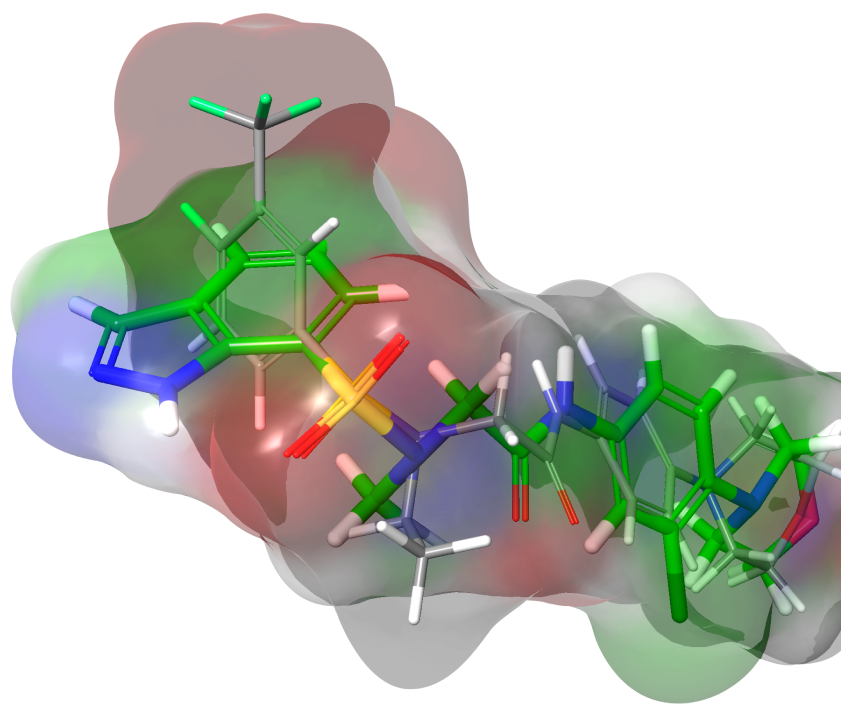
Shape Screening is a ligand-based workflow for efficiently screening ultra-large purchasable or synthesizable compound libraries. Chemically-intuitive ligand alignments are scored based on the quality of the shape overlap against a reference ligand. Shape Screening identifies potential hit molecules with activity even when they are topologically dissimilar to the known reference ligand, making it a powerful ligand-based screening method.
Store and screen tens to hundreds of billions of molecules at speeds exceeding Shape GPU performance by 30% as well as reducing hard disk requirements by a factor of 100
Perform GPU-accelerated high-throughput virtual screenings on millions of molecules with ~11,000 comparisons per second
Screen hundreds of thousands to millions of molecules quickly on multiple CPUs
| Workflow | Description | Library Size | Time to screen 4.0B (days) | Time to screen 6.5B (days) | Storage space for 6.5B (TB) |
|---|---|---|---|---|---|
| Quick Shape | Combination of 1D-SIM* prefilter and Shape CPU Screening | > 4.0 billion | 5.2 | 5.5‡ | 0.4 |
| Shape GPU | GPU-accelerated 3D screening | < 5.0 billion | 4.6 | 7.5‡ | 33 |
| Shape CPU | CPU-based 3D screening | < 10 million | NA | NA | NA |
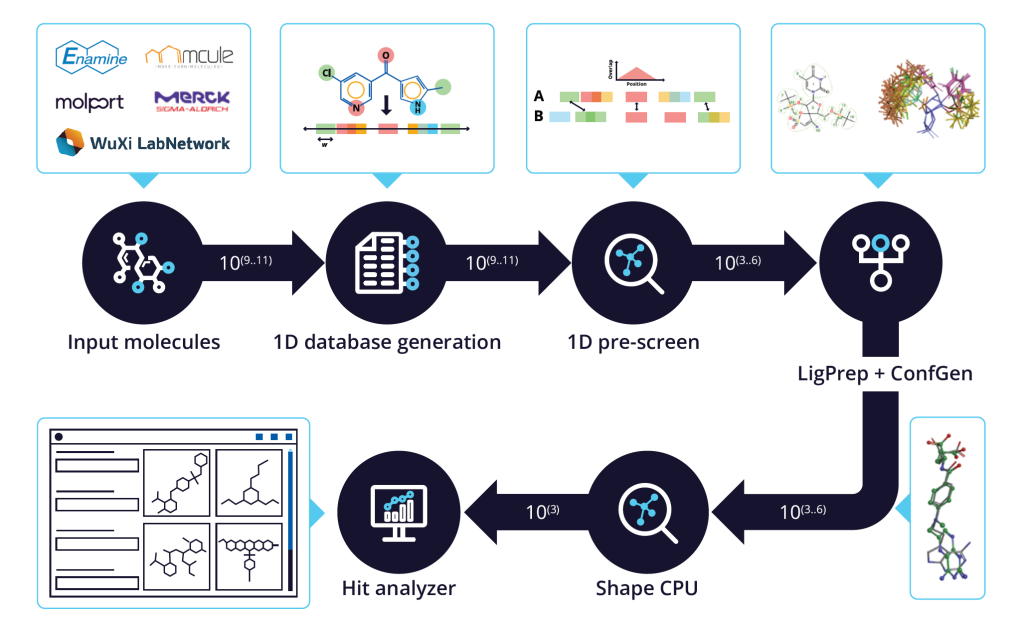
Learn about QuickShape, an automated staged workflow combining both 1D and 3D approaches, which enables efficient 3D-Shape screenings for library sizes in the tens of billions of molecules.
Schrödinger has partnered with leading providers to help you access commercial databases of fragments, lead-like, near drug-like, and drug-like compounds ranging from millions to billions of compounds encompassing a vast chemical space.
Get answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
An easy-to-use pharmacophore modeling solution for ligand- and structure-based drug design
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.