
Case Studies
 Case Study
Life Science
Materials Science
Case Study
Life Science
Materials Science
 Case Study
Life Science
Case Study
Life Science
 Case Study
Life Science
Case Study
Life Science
Documentation
- Documentation
LigPrep
Rapidly prepare high-quality small molecule ligand structures for structure-based virtual screening and other computational workflows.
- Documentation
Ligand Designer
Interactively design a ligand in the context of a protein or DNA/RNA receptor to optimize its binding and properties.
- Documentation
KNIME Extensions
Schrödinger KNIME extensions of more than 160 nodes and provides access to a wealth of ligand- and structure-based tools from the Schrödinger Suite.
Events
 Event
Materials Science
Event
Materials Science
- Jul 8, 2025
Digital and AI-Driven Materials Innovation
We hope you can join us at The Hong Kong University of Science and Technology (HKUST) in Hong Kong, on July 8th for the Digital and AI-Driven Materials Innovation Symposium.
 Event
Materials Science
Event
Materials Science
- Jul 13th-16th, 2025
IFT First Annual Event and Expo 2025
Schrödinger is excited to be participating in the IFT First Annual Event and Expo 2025 conference taking place on June 13th – 16th in Chicago, Illinois.
 Event
Materials Science
Event
Materials Science
- Jul 14th-18th, 2025
CRS 2025
Schrödinger is excited to be participating in the CRS Annual Meeting 2025 taking place on July 14th – 18th in Philadelphia, Pennsylvania.
Training Videos
 Video
Life Science
Video
Life Science
Getting Going with Maestro BioLuminate
A free video series introducing the basics of using Maestro Bioluminate.
 Video
Life Science
Video
Life Science
- Video
Introducing Ligand Designer
An overview of the LigandDesigner workflow, Editing in 2D and 3D, using display options and overlays, and accessing the Admin Panel.
Publications
- Publication
- May 9, 2025
Efficient long-range machine learning force fields for liquid and materials properties
Weber JL, et al. arXiv, 2025, Preprint- Publication
- Apr 18, 2025
Enabling in-silico Hit Discovery Workflows Targeting RNA with Small Molecules
Chopra, et al. Theoretical and Computational Chemistry, 2025, 1, Preprint- Publication
- Apr 17, 2025
Active Learning FEP: Impact on Performance of AL Protocol and Chemical Diversity
Lonsdale, et al. Journal of Chemical Theory and Computation, 2025Quick Reference Sheets
- Quick Reference Sheet
Synthesis Queue LiveReport
Use Freeform columns to track the status of compounds in a synthesis queue.
- Quick Reference Sheet
Modeling Queue LiveReport
Learn how to use Freeform columns and an Auto-Update Search to create compound progression workflows.
- Quick Reference Sheet
Group Meeting LiveReport
Develop a workflow to allow individuals to triage ideas for group discussion and decision making.
Tutorials
- Tutorial
Structure-Based Virtual Screening using Glide
Prepare receptor grids for docking, dock molecules and examine the docked poses.
- Tutorial
Ligand Binding Pose Prediction for FEP+ using Core-Constrained Docking
Generate starting poses for FEP simulations for a series of BACE1 inhibitors using core constrained docking.
- Tutorial
Antibody Visualization and Modeling in BioLuminate
Visualize, build, and evaluate antibody models, analyze an antibody for various characteristics, dock an antigen to an antibody.
Webinars
 Webinar
Life Science
Webinar
Life Science
- Jul 16, 2025
Schrödinger デジタル創薬セミナー: Into the Clinic ~計算化学がもたらす創薬プロセスの変貌~ 第18回
Enabling cryoEM structures for drug discovery with the Schrödinger Suite
 Webinar
Life Science
Webinar
Life Science
- Jul 30, 2025
In silico cryptic binding site detection and prioritization
In this webinar, we will introduce a novel computational workflow that integrates mixed solvent molecular dynamics (MxMD) with SiteMap to reveal and identify cryptic binding sites.
 Webinar
Life Science
Webinar
Life Science
- Jun 25, 2025
How to find a druggable target: A computational perspective
Join us in this beginner-friendly webinar that will introduce you to strategies and best-in-class tools for identifying druggable, technology-enabled targets.
White Papers
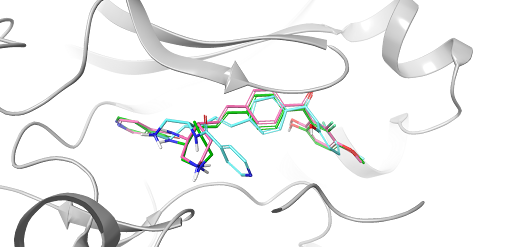 White Paper
Life Science
White Paper
Life Science
- Oct 29, 2024
20 Years of Glide: A Legacy of Docking Innovation and the Next Frontier with Glide WS
Glide has long set the gold standard for commercial molecular docking software due to its robust performance in both binding mode prediction and empirical scoring tasks, ease of use, and tight integration with Schrödinger’s Maestro interface and molecular discovery workflows.
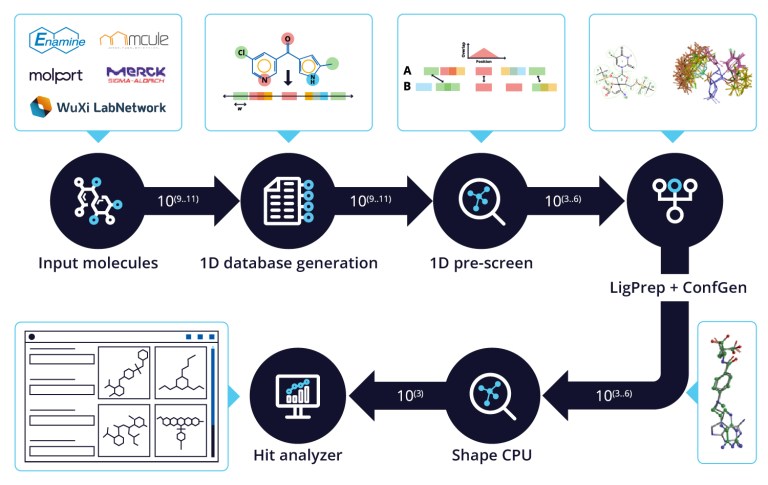 White Paper
Life Science
White Paper
Life Science
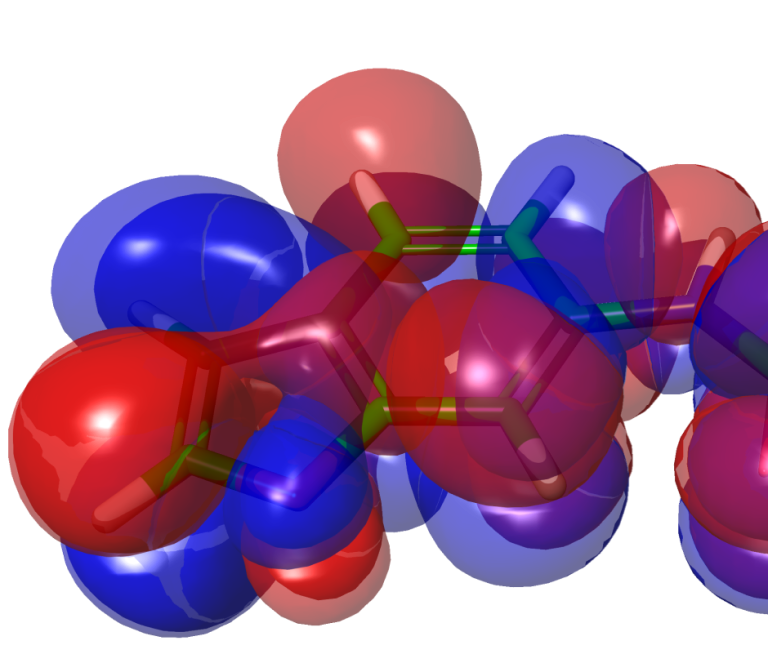 White Paper
Life Science
White Paper
Life Science

Latest insights from Extrapolations blog

With FEP+, “The Experiment is the Limit.”
Over the past century, small molecule drugs have represented the dominant modality in drug research, enabling medical breakthroughs that have saved countless lives.

Tackling Drug Solubility: AbbVie and Schrödinger Collaborate to Advance Accurate Prediction Methods
The complexity and size of drug candidates has grown in recent years as scientists pursue novel targets once considered undruggable.
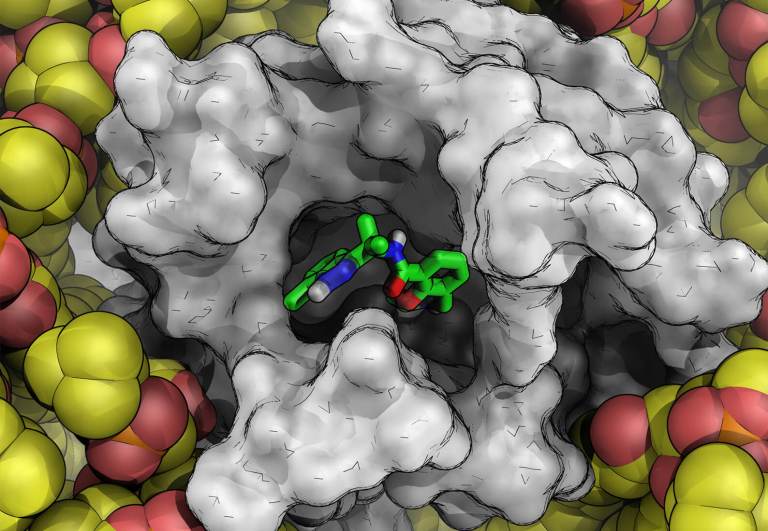 Blog
Blog
Can AlphaFold Models be Used for Structure-Based Drug Design? A Perspective Two Years In
We recently sat down with Edward Miller, Senior Director of Protein Structure Modeling at Schrödinger, to discuss his experience using AlphaFold models for SBDD.
Training & Resources
Online certification courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Free learning resources
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Other Resources