OCT 29, 2025
Advancing battery materials innovation using charge-aware machine learning force fields
Batteries are fundamental technology – powering everything from our personal electronics to electric vehicles, as well as large-scale grid storage systems for renewable energy integration. However, current battery technologies, primarily lithium-ion batteries, face significant limitations in performance, safety, cost, and reliance on scarce materials like cobalt. Therefore, innovation in battery materials is the key to unlocking the next generation of energy storage.
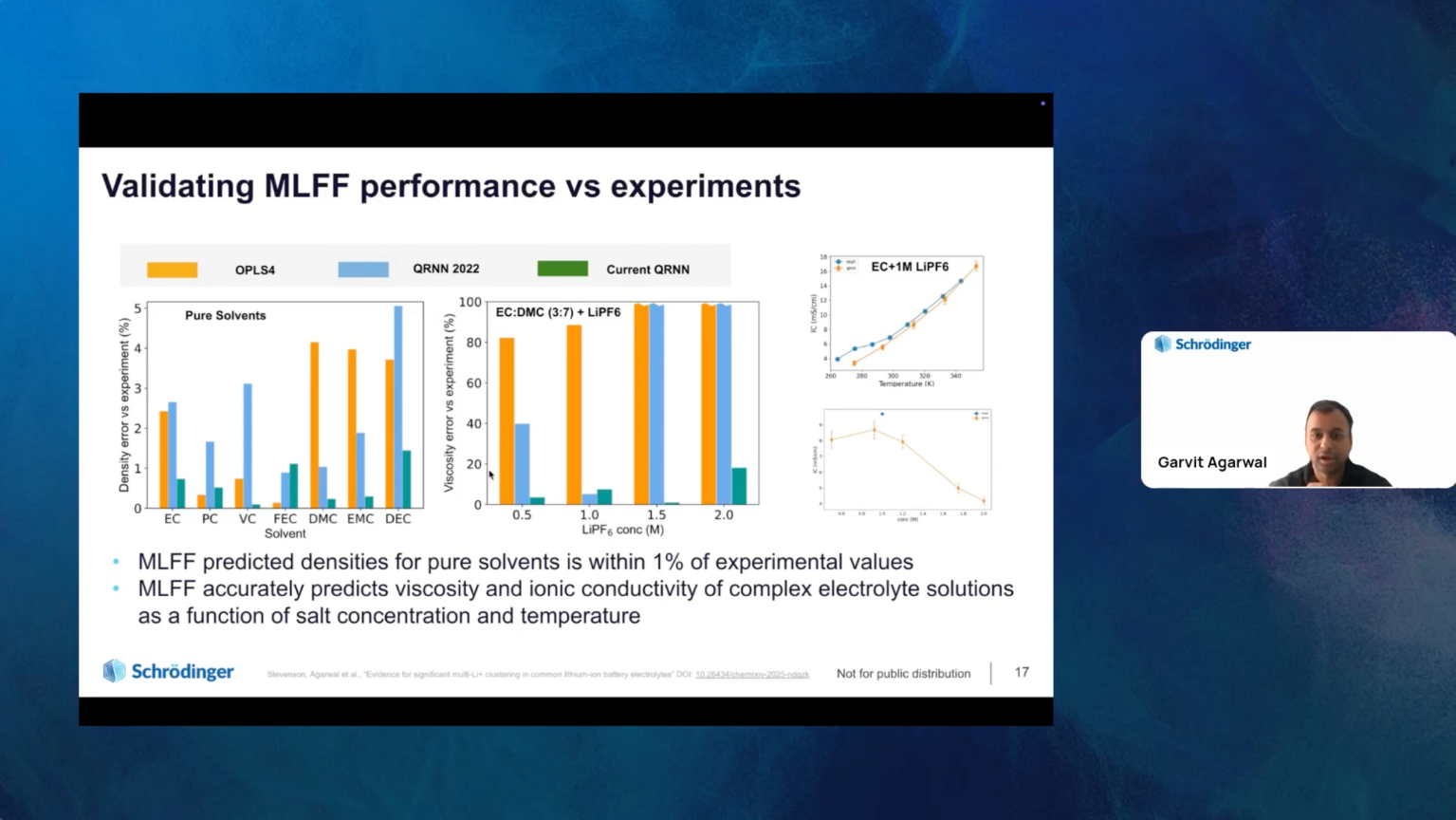
In this webinar, we will demonstrate how Schrödinger is utilizing an integrated computational approach combining physics-based molecular modeling with machine learning force fields (MLFFs) to address key challenges in battery materials design. We will introduce Schrödinger’s latest advancements in MLFFs, featuring charge recursive neural networks (QRNN) and the recently released Message Passing Network with Iterative Charge Equilibration (MPNICE) architectures, which incorporate explicit electrostatics for accurate charge representations.
Moreover, we will showcase several industry-relevant case studies highlighting the application of MLFFs to precisely model the structure and properties of electrolyte materials (liquid, polymer, and inorganic solid-state electrolytes), cathode coatings, and electrode materials. We will also explore how MLFFs facilitate large-scale simulations, allowing scientists to investigate the impact of defects and heterogeneities on crucial properties like Li-ion transport, paving the way for the efficient design of next-generation battery materials and chemistries.
Webinar Highlights:
- How Schrödinger combines physics-based modeling with machine learning force fields to drive battery materials discovery
- Schrödinger’s latest MLFF technologies, including QRNN and MPNICE
- Real-world case studies modeling electrolytes, cathode coatings, and electrode materials
- How MLFFs facilitate large-scale simulations, such as the investigation of Li-ion transport
Our Speaker

Garvit Agarwal
Principal Scientist, Schrödinger
Garvit Agarwal, Principal Scientist and Scientific Lead for Energy Storage at Schrödinger, works to extend and apply molecular modeling tools for the accelerated discovery of next-generation clean energy technologies. Garvit obtained his Ph.D. in Materials Science and Engineering from the University of Connecticut. He worked as a post-doctoral researcher in the Materials Science Division at Argonne National Laboratory prior to joining the Materials Science team at Schrödinger.