Abstract:
Background & Research Question
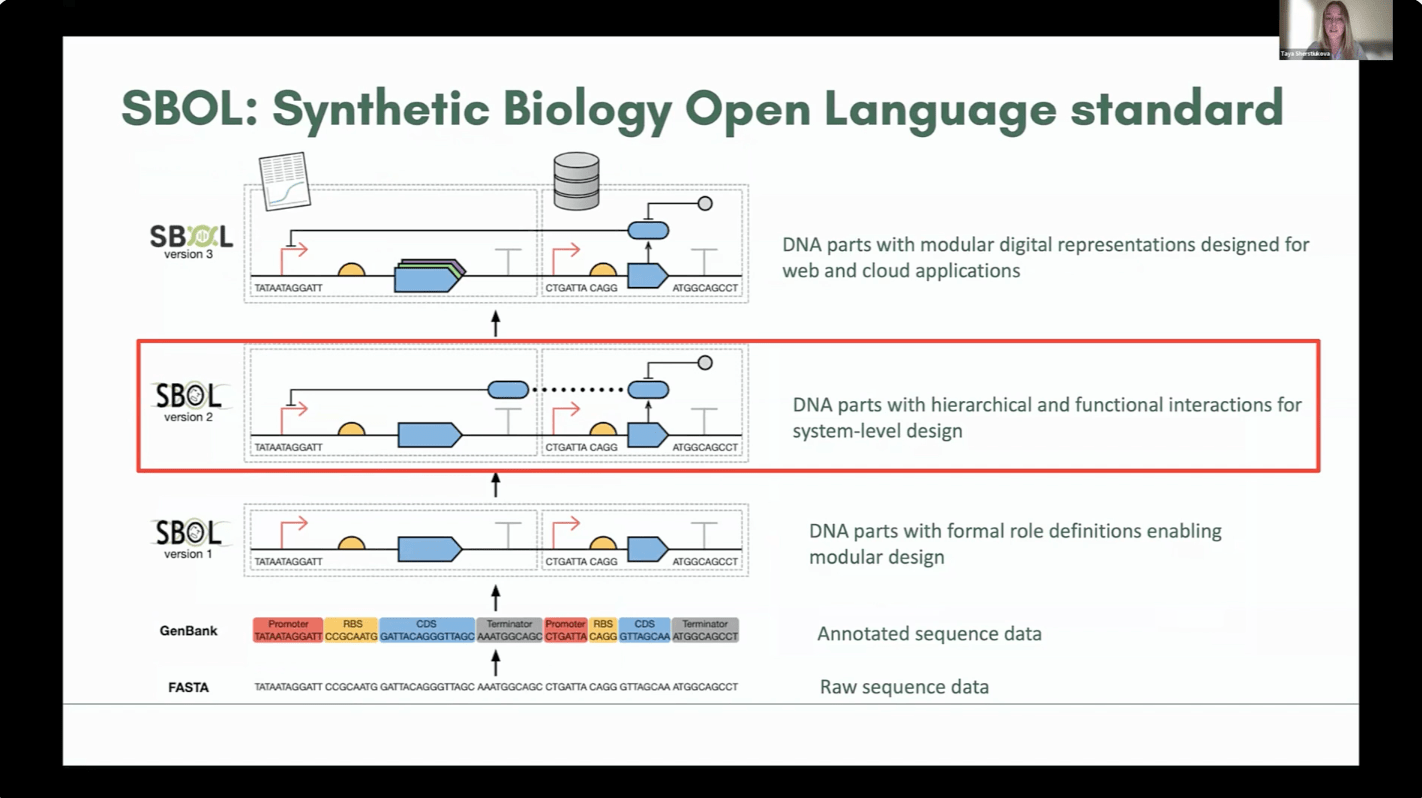
Synthetic biology relies on data standardization to support collaboration and reuse. SBOL (Synthetic Biology Open Language) is a key standard but has a steep learning curve, especially for biologists with limited programming experience. This project extends an Excel-to-SBOL converter tool, enabling users to generate SBOL-compliant files from familiar spreadsheet formats to improve SBOL adoption and streamline data sharing.
Methods
Using a Python script and, in particular, the pySBOL2 library, this project translates biological design elements from spreadsheet rows into structured SBOL objects. The tool supports key design patterns such as transcriptional regulation, protein production, and complex formation by generating corresponding ComponentDefinition and ModuleDefinition objects with appropriate interactions (e.g., inhibition, stimulation, binding). Each interaction, functional components, and ontological annotations are added to an SBOL document. The output is validated and uploaded to SynBioHub to ensure structural integrity and enable visual exploration.
Results
As a result of this project, I successfully implemented new features for modeling genetic production, repression, activation, biochemical reaction, DNA and protein sequencing, and complex component formation. In addition, I deployed sequence and part name validation to detect duplicates and ensure structural data integrity. These contributions expanded the tool’s ability to capture complex biological interactions from spreadsheets and improved the reliability of SBOL output, supporting more accurate and reusable designs for synthetic biology workflows.
Conclusion & Implications
By lowering the barrier to SBOL generation, Excel-to-SBOL supports broader adoption of standardized design practices in synthetic biology. Available as a Python package, it is already used by research labs and organizations nationwide, facilitating reproducibility and accelerating collaborative design efforts. Future work will ensure compatibility and conversion with SBOL3 data format and expand functionality to support a broader range of biological interactions as user needs evolve.
Speaker:
Taisiia Sherstiukova, University of Colorado Boulder
Taisiia Sherstiukova is a recent graduate from the University of Colorado Boulder with a Bachelor’s degree in Computer Science. Her work focuses on simplifying synthetic biology workflows by developing tools that bridge the gap between lab scientists and computational tools.