JUN 12, 2025
Analyzing Quantum Exceptional Point Invisibility for Experimentally Realizable Triple-Gaussian Potentials
Abstract:
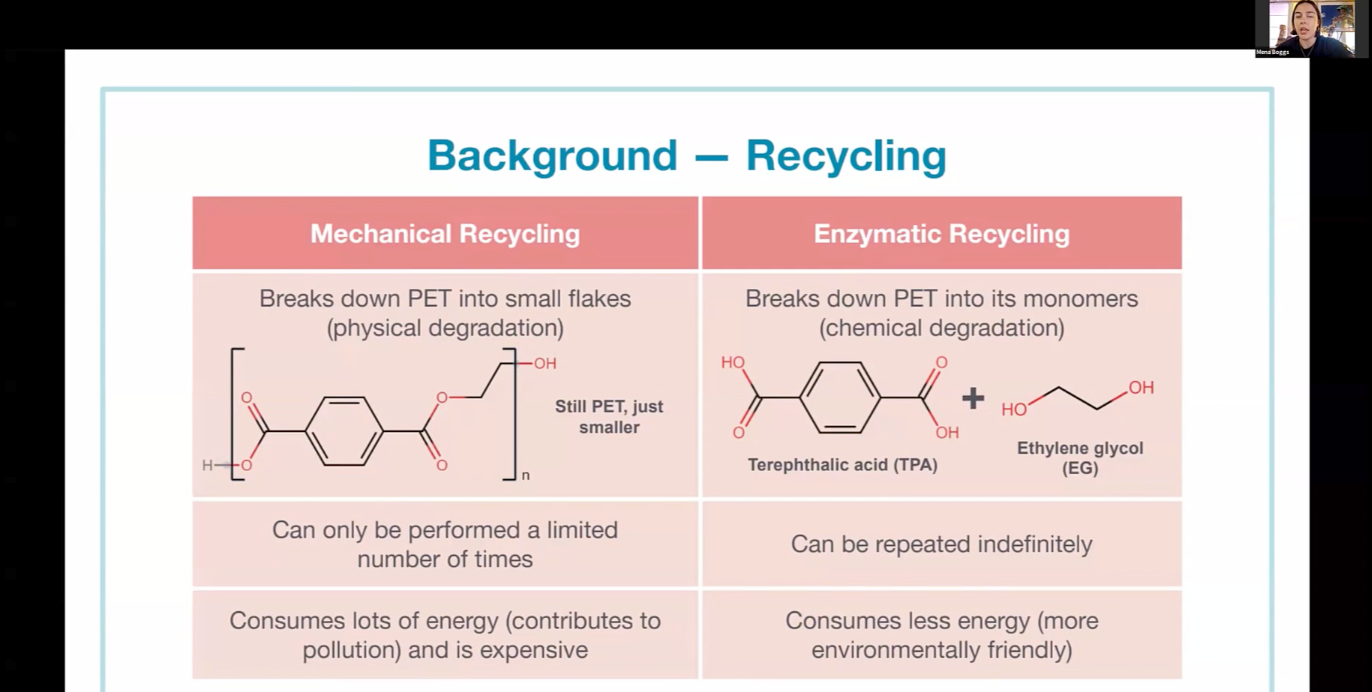
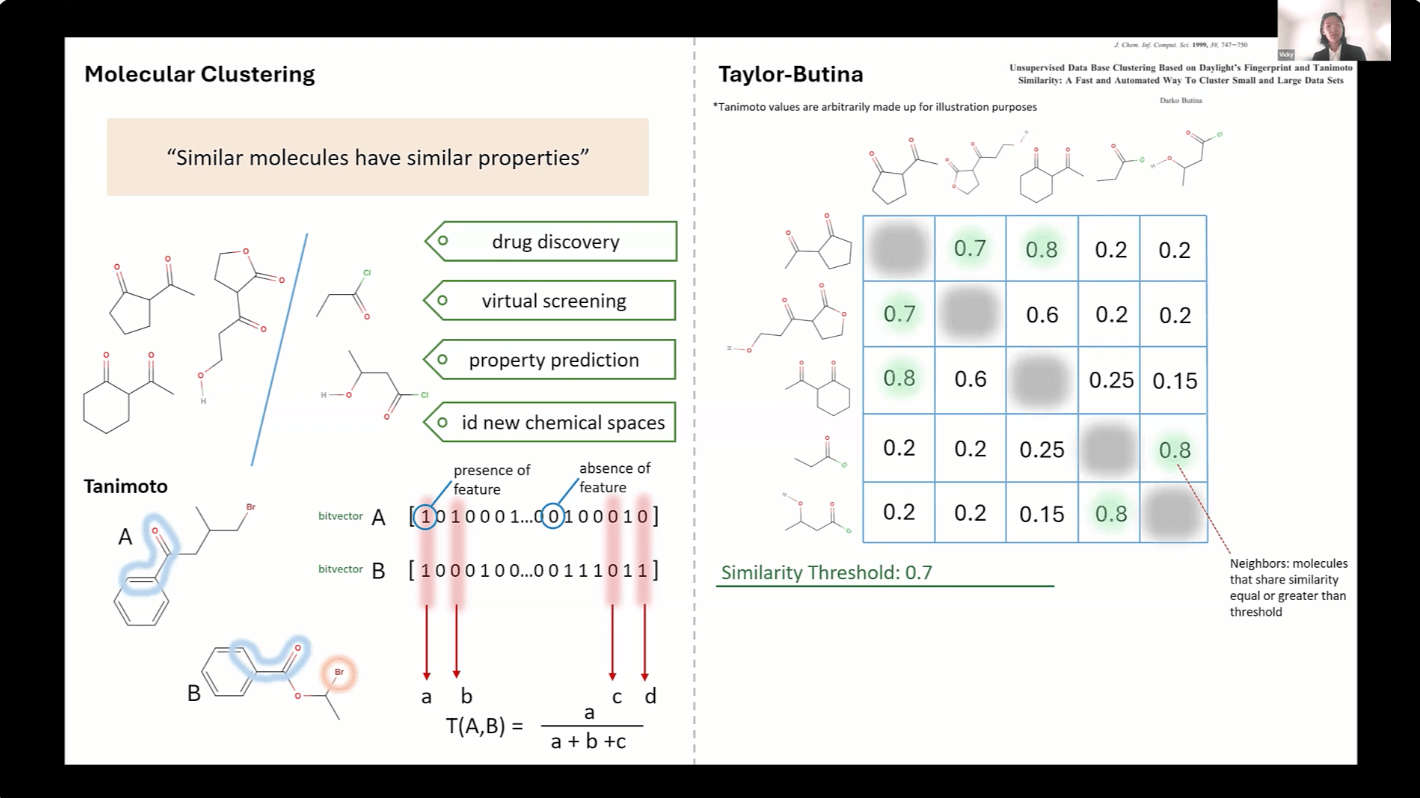
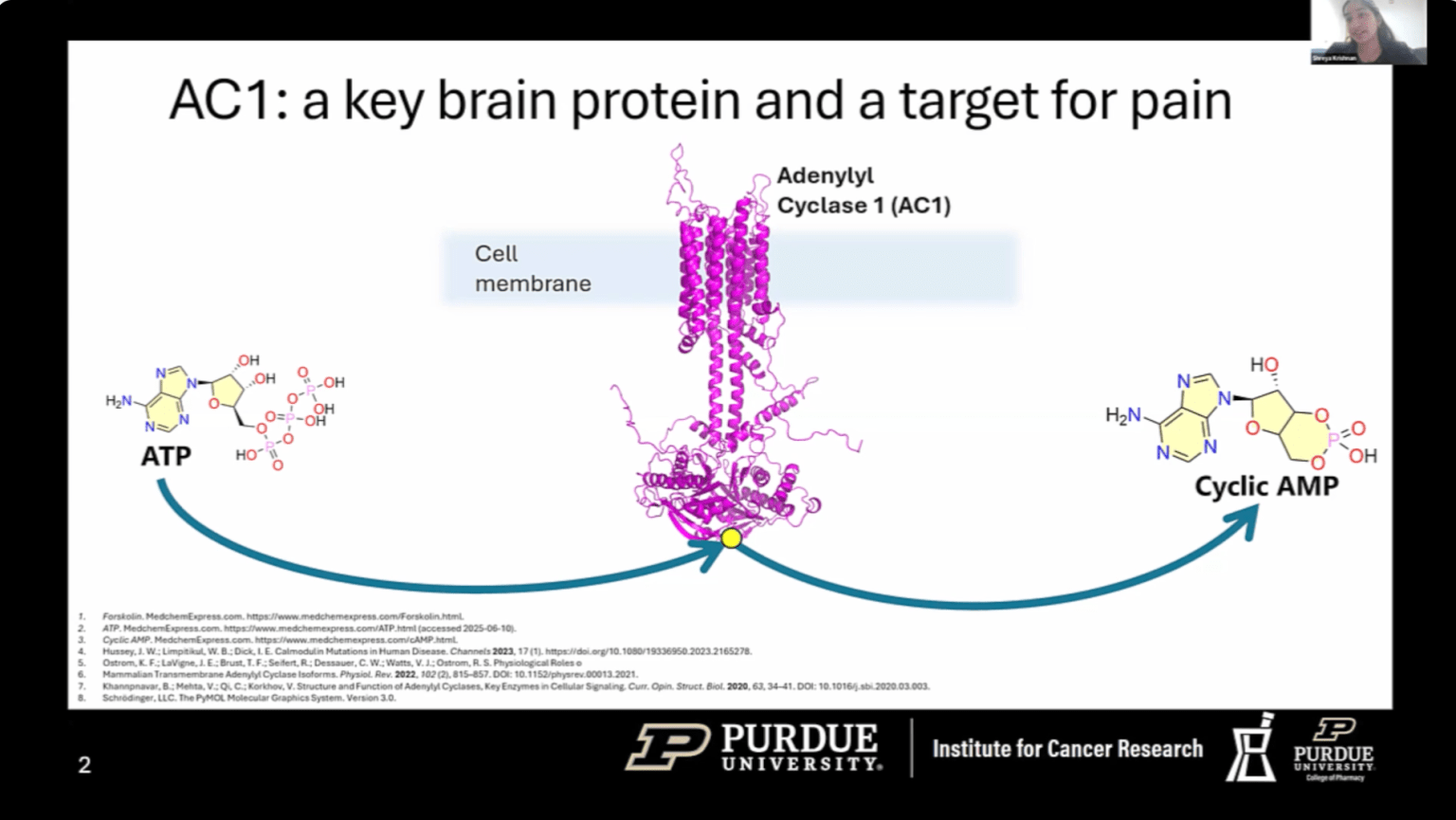
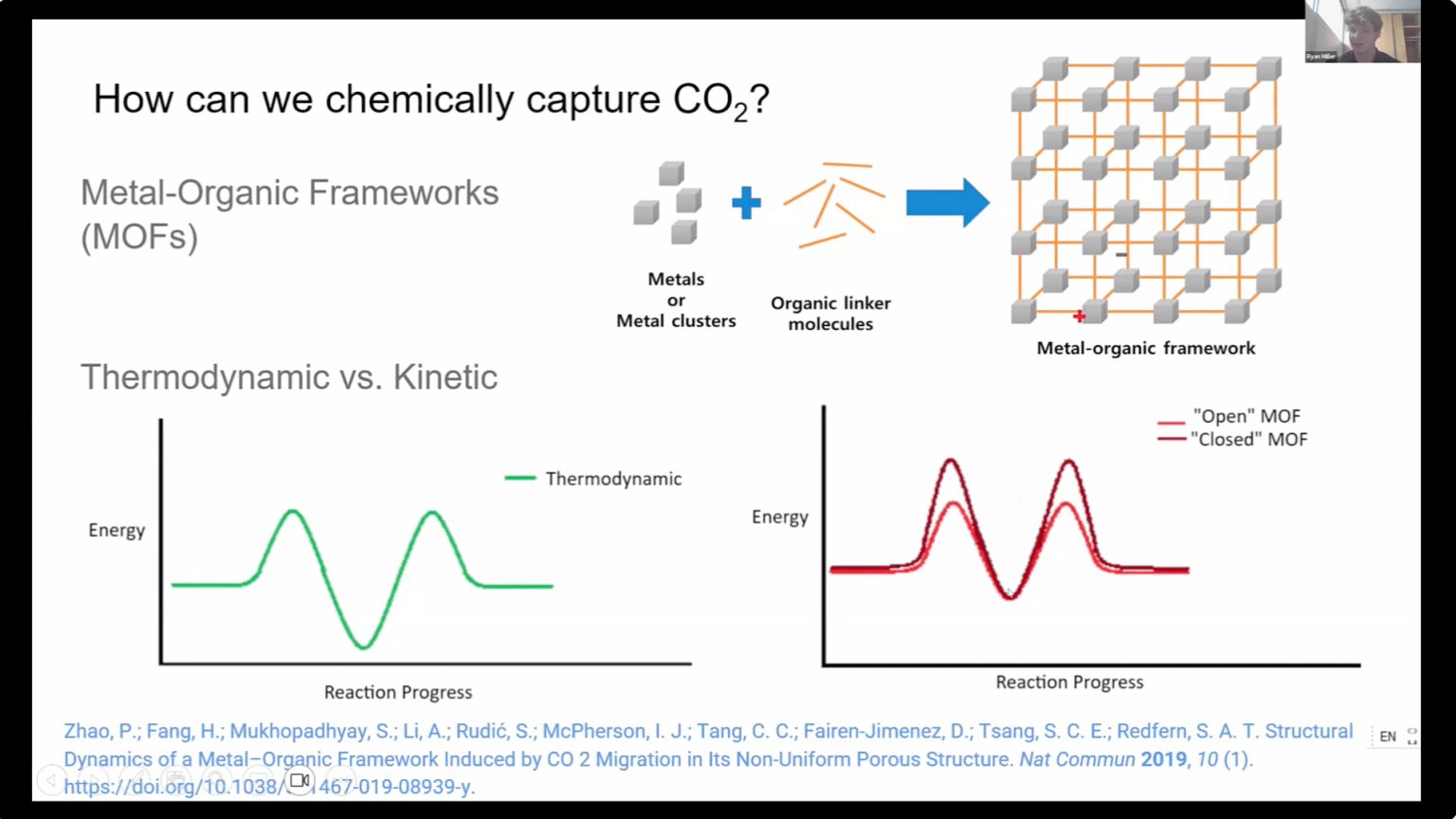
Quantum sensing leverages the extreme sensitivity of quantum systems like ultracold atoms and molecules to probe minute changes in their environment beyond classical limits. This sensitivity becomes especially pronounced near exceptional points (EPs)–critical parameter regimes where eigenstates coalesce to produce nonlinear responses, nonreciprocal dynamics, and phase transitions. For instance, quantum scattering EPs correspond to invisibility points where particles pass through barriers unimpeded. We aim to advance high-precision quantum molecular sensors utilizing these properties by establishing experimentally realizable EPs for quantum particles interacting with one-dimensional potential barriers. We analyze the triple-Gaussian potential due to its parity-time reversal-symmetry phase transition and simple optical implementation. Particle data is computed by numerically solving Schrödingerʼs equation and benchmarked with the triple-rectangular potential using the analytic Transfer Matrix Method. We demonstrate theoretically the successful realization of EP invisibility effects with a triple-Gaussian potential, verifiable with an ultracold atom experiment using rubidium-87 Bose-Einstein condensates and spatial light modulators. A key finding is that raising the central Gaussian barrier out of the three merges EPs, acting as a quantum filter by selectively permitting certain energies to pass. Additionally, we discover novel EPs for the triple-delta potential, a theoretical system consisting of three infinitesimally thin barriers acting as a direct atomic or molecular analog of optical systems currently under investigation for high-precision quantum sensors. Understanding EPs has broad applications, including sensitive quantum sensors for early earthquake detection, noninvasive biosensing through “invisible” nanoparticles for effective diagnostics and treatments, and enantio-sensitive quantum control for chemical reactivity. This project lays the foundation for further EP studies, with future work incorporating thermal noise to support experimental testing and modeling three-dimensional systems to develop practical atomic and molecular quantum technology.
Speaker:
Shrikar Dulam, University of Illinois, Urbana-Champaign
Shrikar Dulam is an undergraduate student at the University of Illinois Urbana-Champaign, pursuing a degree in Computer Science + Physics. His research interests lie at the intersection of quantum physics and computational science, with a focus on quantum computing and quantum sensing.