Accessible and automated computational catalyst discovery and reactivity optimization
- Virtual
Designing high-performing homogeneous catalysts and optimizing non-catalytic transformations often relies on trial-and-error iteration. On the other hand, computational tools often remain inaccessible to those without specialized expertise.
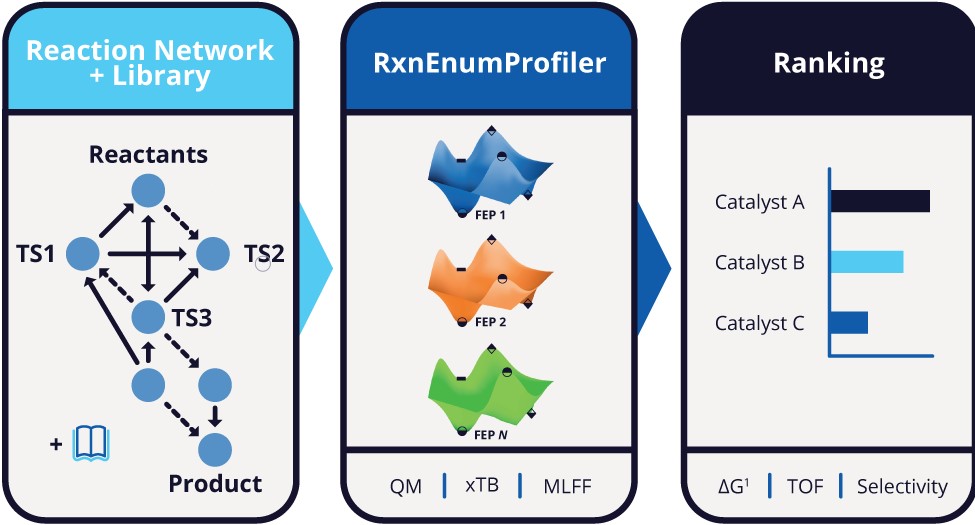
In this webinar, we will demonstrate how an end-user physics–AI platform removes barriers to entry, making this process accessible to both experts and non-experts while enabling seamless scalability. By integrating quantum chemistry, fast semiempirical tight-binding methods, and machine learning interatomic potentials, the platform enables automated exploration of reaction networks and simultaneous optimization of multiple performance metrics. Using real-world catalytic and non-catalytic examples, we will show how mechanistic insight can be translated into actionable design decisions—quickly, accurately, and without coding.
Key highlights:
- Lower the barrier to catalyst design
Discover how a Physics–AI end-user platform enables both experts and non-experts to perform advanced in silico catalyst design and reactions optimization - Multi-level modeling at scale
Leverage quantum chemistry, semiempirical tight-binding methods, and machine learning interatomic potentials (MLIPs) within a single, scalable framework - No coding required
Intuitive, GUI-driven environment designed for accessibility and productivity
Who should attend: Anyone interested in non-catalytic and catalytic reactivity optimization, or homogeneous catalyst design
Our Speaker

Pavel Dub
Research Leader and Product Manager, Catalysis & Reactivity, Schrödinger
Pavel A. Dub earned a Ph.D. in Physical Chemistry from the A. N. Nesmeyanov Institute of Organoelement Compounds and a second Ph.D. from the Université de Toulouse. He subsequently completed postdoctoral appointments at the Tokyo Institute of Technology and Los Alamos National Laboratory, where he later served as a Staff Scientist. In 2022, he joined Schrödinger. His research focuses on computational chemistry and materials science across both classical and quantum computing architectures.