EFMC Medicinal Chemistry 2026
- September 6th-10th, 2026
- Basel, Switzerland
Schrödinger is excited to be participating in the EFMC Medicinal Chemistry 2026 conference taking place on September 6th – 10th in Basel, Switzerland. Join us for a workshop by David Rinaldo, Senior Principal Scientist, Applications Science at Schrödinger, titled “Accelerating Drug Discovery with Integrated AI/ML Modeling.”
Accelerating Drug Discovery with Integrated AI/ML Modeling
Speaker:
David Rinaldo, Senior Principal Scientist, Applications Science at Schrödinger
Abstract:
AI/ML models are now indispensable in modern drug discovery, offering powerful
capabilities ranging from protein structures predictions to ligand property prediction and
including 3D protein-ligand binding pose prediction or de novo molecular design.
However, effectively deploying and managing these models requires a centralized,
collaborative platform.
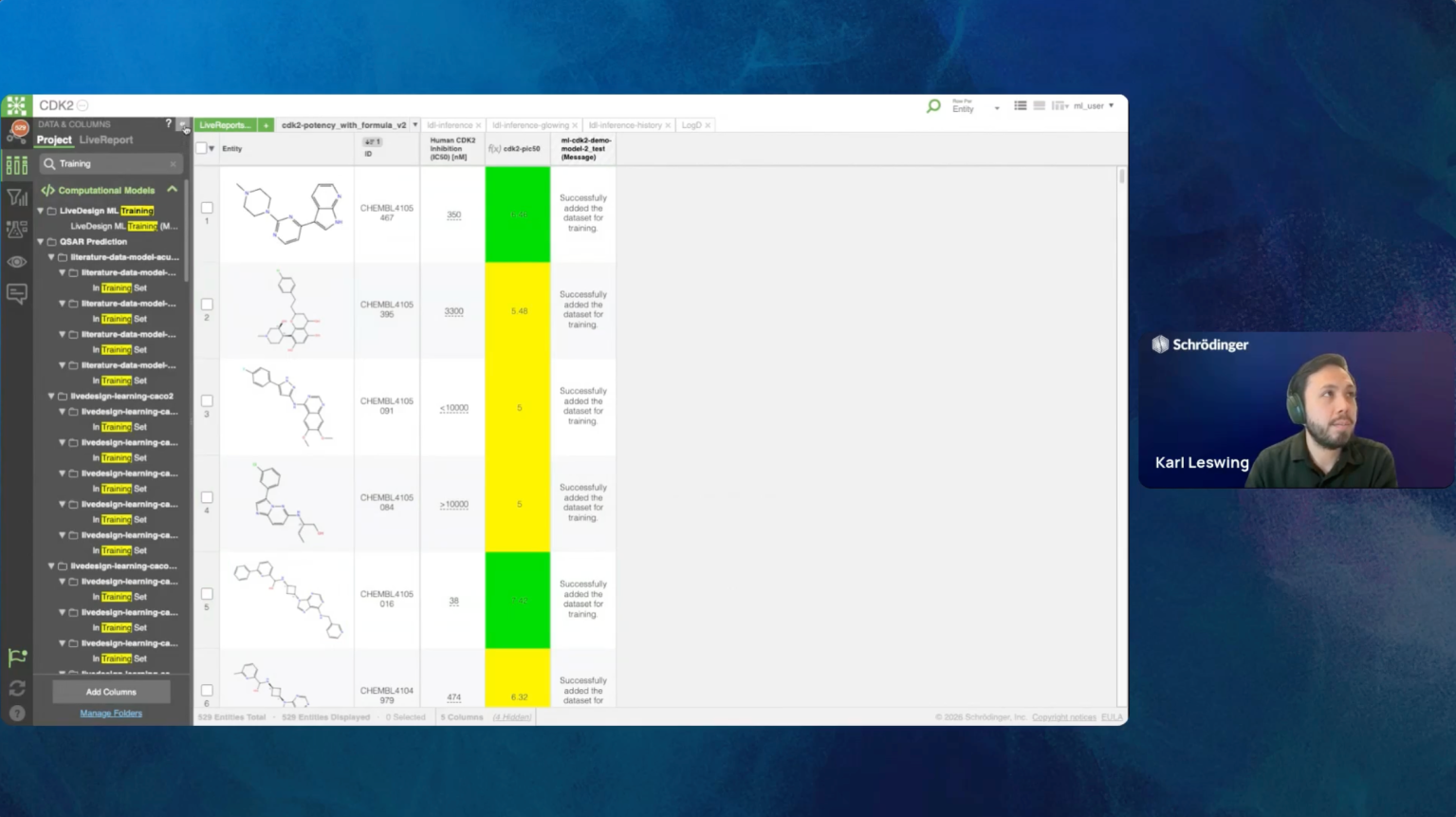
LiveDesign-ML is the module within the LiveDesign platform that empowers scientists to
generate, validate, and deploy state-of-the-art AI/ML models with minimal manual
intervention. We will demonstrate its capability for molecular property predictions, which
are crucial for triaging newly designed ideas and enabling the screening of hundreds of
thousands of compound ideas in minutes. By treating datasets as dynamic information
feeds, LiveDesign ML ensures models are always optimized and reliable for your
evolving chemistry.
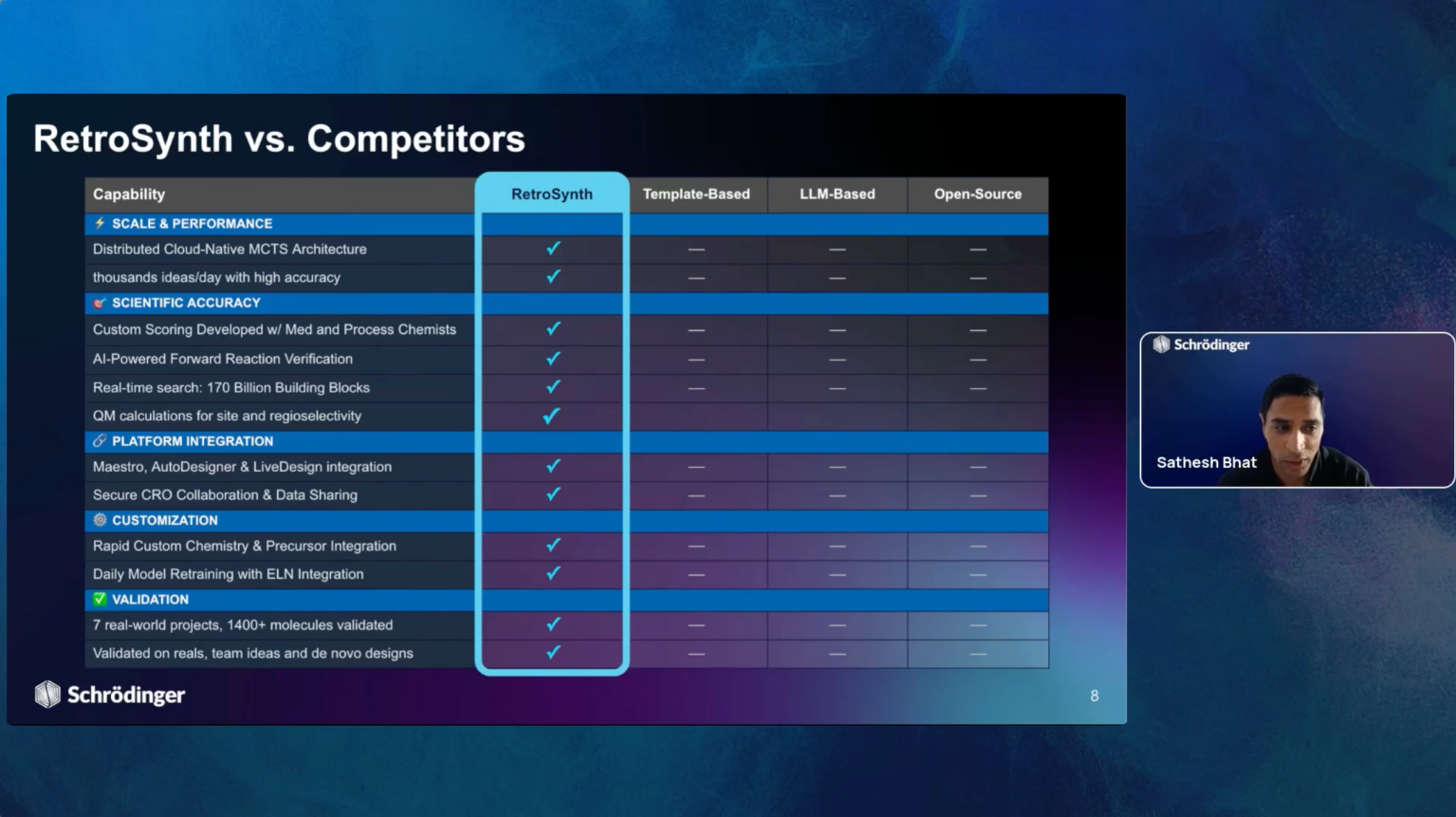
We will also introduce RetroSynth, Schrödinger’s AI-driven synthesis planning platform.
Engineered to accelerate and scale conventional retrosynthesis, RetroSynth uses
advanced deep learning and a cloud-native Monte Carlo tree search (MCTS)
architecture to predict and score optimal, accurate, and cost-efficient synthetic
pathways. Learn how the integration of real-time building block data with AI and
physics-based modeling in RetroSynth unlocks accurate retrosynthetic analysis, leading
to massive project acceleration and significant cost savings in hit identification and lead optimization.
Medicinal chemists, computational chemists, and R&D leaders are welcome to join this
workshop. We will showcase how Schrödinger’s LiveDesign-ML and RetroSynth are
integrated to tackle critical challenges in the design and synthesis workflow. It will also
be the opportunity to see the full potential of integrated AI/ML and physics-based
modeling to overcome bottlenecks and advance your drug discovery programs.