Advancing machine learning force fields for materials science applications

AUG 6, 2025
Advancing machine learning force fields for materials science applications
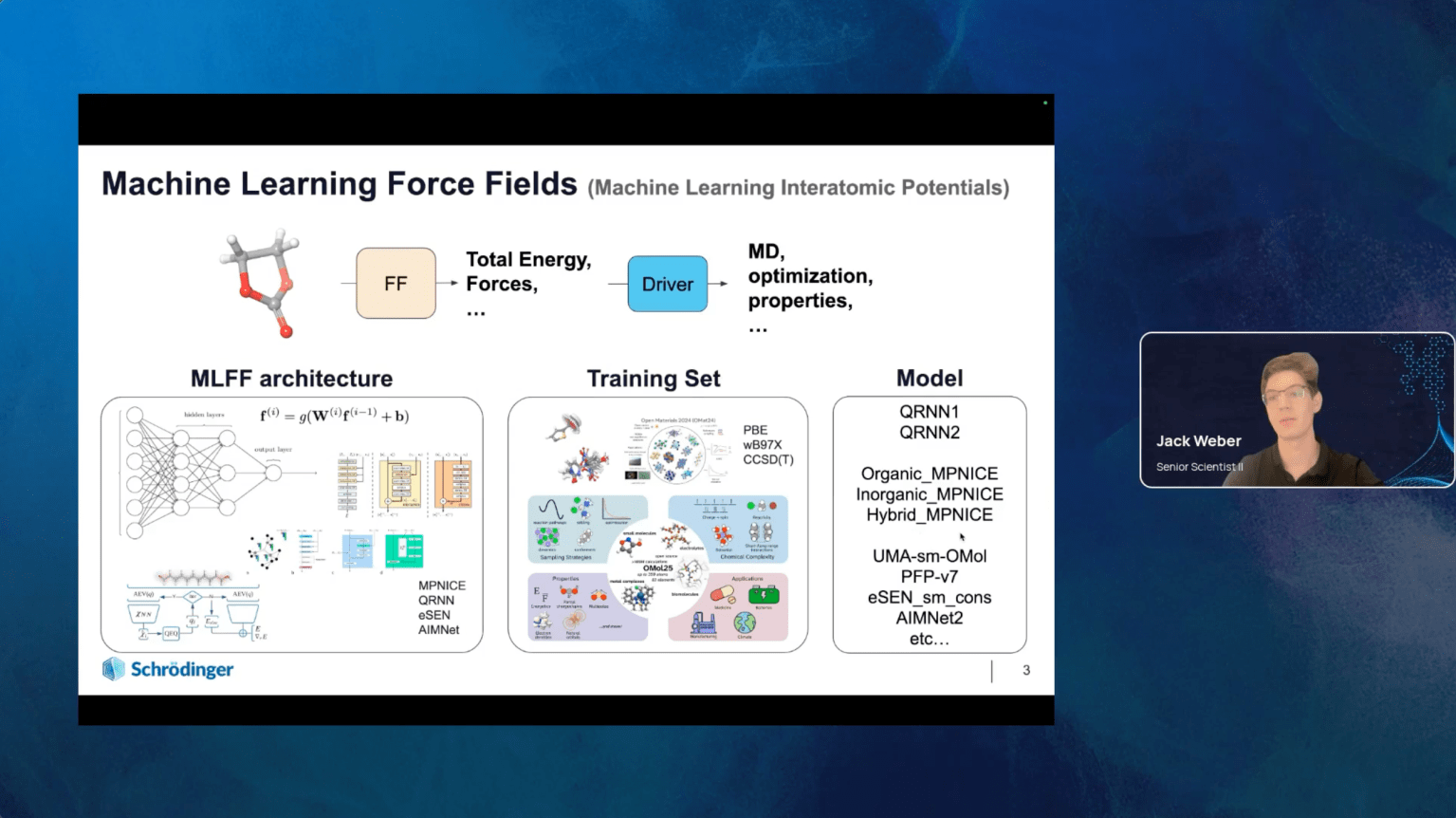
Machine learning force fields (MLFFs), also referred to as machine learning interatomic potentials, have emerged as a critical tool for the cost-efficient atomistic simulations of diverse chemical systems, often achieving density functional theory (DFT) accuracy at a fraction of the cost. Recent advances in message passing networks have removed the drawback of previous MLFFs that were limited by the number of unique atomic elements they could model. Furthermore, inclusion of atomic charges and electrostatics through charge equilibration approaches have enabled accurate representations of multiple charge states, ionic systems, and electronic response properties, while simultaneously improving accuracy using explicit long-range interactions.
In this webinar, we will introduce Schrödinger’s state-of-the-art MLFF architecture, called Message Passing Network with Iterative Charge Equilibration (MPNICE), which incorporates explicit electrostatics for accurate charge representations. We present a family of pre-trained models trained on materials covering the entire periodic table (89 elements). MPNICE prioritizes efficient throughput, enabling atomistic simulations at length and time scales that were previously inaccessible without sacrificing accuracy. We will outline available tools in the Schrödinger suite that incorporate MLFFs to enable larger and more complex simulations for materials design, providing industry relevant examples throughout.
Highlights:
- Overview of MPNICE – a message passing MLFF architecture that includes atomic partial charges and long-range interactions, while maintaining speeds an order of magnitude faster than comparable models
- Highlights of recent applications of MPNICE, including general organic, inorganic, and hybrid (organic and inorganic) models to address industry relevant needs
Our Speaker

Jack Weber
Senior Scientist, Schrödinger
Jack Weber is a Senior Scientist at Schrödinger, where he develops machine learning force fields (MLFFs) for applications in drug discovery and materials science. Jack received his PhD in Chemical Physics in 2023 from Columbia University, advised by Professors Richard Friesner and David Reichman. In his doctoral research, he used advanced computational methods to investigate fundamental problems in chemistry and materials science, including improving ab-initio methods in electronic structure to treat strongly correlated systems.
Leveraging machine learning applications combined with physics-based modeling for drug discovery


OCT 4, 2023
Leveraging machine learning applications combined with physics-based modeling for drug discovery
Abstract:
Machine learning strategies in drug discovery are becoming increasingly popular and can be used in various areas. In the Schrödinger Suite DeepAutoQSAR serves as the main tool for training machine learning models to predict activity, ADMET, and other compound properties. In order to leverage both the proven accuracy and wide applicability domain of physics-based computational models, such as QM and FEP, together with the speed and scale of machine learning, we have combined our physics-based modeling technologies with an active learning framework. This framework can effectively speed up virtual screening methods such as in Active Learning -Glide, Active Learning-FEP, and Active Learning-ABFEP, or to improve the accuracy and applicability domain of models such as pKa prediction in Epik and machine learned force fields such as QRNN. We will also discuss how to utilize machine learning protein structure prediction methods to enable new targets for structure-based drug design.
Speaker:
Dr. Marton Vass, Principal Scientist II, Schrödinger
Virtual screening with integrated physics & machine learning

Virtual screening with integrated physics & machine learning
Acquire essential skills in next-generation virtual screening, integrating physics and machine learning for smarter hit identification
Details
Available Languages
Chinese, English, Japanese, Korean
Duration
5 weeks from selected start date
Level
Intermediate
Cost
$715 for non-student users
$270 for student / post-doc
Who should take this course?
Medicinal chemists, cheminformaticians, ML scientists, new computational chemists
Overview
With the growing demand for computationally efficient methods to explore chemical space, machine learning-enabled virtual screening has become a critical tool in modern drug discovery. These techniques empower chemists to rapidly identify and prioritize high-potential hits and chemotypes for experimental validation and downstream development.
Schrödinger’s online course, Virtual screening with integrated physics and machine learning, offers expert instruction and best practices for executing these workflows from start to finish.
Ideal for scientists looking to enhance their practical skills and deepen their understanding of both ligand-based and structure-based approaches, this course also provides a certification and digital badge to showcase your expertise in this cutting-edge field.
- Accelerate your research through target-diverse hands-on exercises with Schrödinger’s industry-leading Maestro and command line interface.
- Execute complete, next-generation drug discovery workflows, from initial preparation to large-scale data analysis and informed decision-making.
- Scale virtual screening workflows using Active Learning Glide.
- Score compounds with Glide WS and analyze binding affinity data from Free Energy Perturbation (FEP+) calculations.
- Apply your learning in a comprehensive, real-world case study, showcasing your readiness to tackle advanced virtual screening challenges.
- Benefit from personalized review and feedback from Schrödinger Education Team experts for course assignments and related queries.
This course comes with temporary access to a web-based version of Schrödinger software, complete with licenses and compute resources
Requirements
- A computer with reliable high speed internet access (8 Mbps or better)
- A mouse and/or external monitor (recommended but not required)
- Working knowledge of general chemistry
- Working knowledge of Maestro. Please work through our Getting Going with Maestro resources to become familiar with using Maestro.
- (Optional) Prior completion of the Designing quality ligand libraries or Target enablement, preparation, & validation online certification courses.
Certification
- A certificate signed by the Schrödinger course lead to add to your CV or resume
- A badge that can be posted to social media, such as LinkedIn

What you will learn
Executing and Validating Virtual Screens
Get hands-on experience with pilot virtual screens, including ligand and receptor grid preparation, and learn critical methods for validating the results of docking simulations
Hit Evaluation and Prioritization
Learn to effectively evaluate and prioritize hits after screening, including inspection, clustering, and scoring
Scaling Workflows with Advanced Methods
Explore how machine learning and Free Energy Perturbation (FEP+) methods can be integrated to scale virtual screening workflows
Practical Application in Drug Discovery
Apply your knowledge through a comprehensive case study, following the end-to-end process of a virtual screen from initial setup to the evaluation of potential drug candidates
Modules
Module 1
2 Hours
Course overview and modern virtual screening methods

Videos
- Course overview
- Meet the instructors

Syllabus and honor code
Expectations surrounding academic integrity

Videos
- In-silico technology at Schrödinger
- Introduction to hit discovery

End of module checkpoint
Module 2
5 Hours + compute time
Executing pilot virtual screens and validating Glide docking results
Videos
- Virtual screening foundations
- Target enablement
- Model validation
- Chemical space

Tutorials:
- Preparing cognate ligands with LigPrep
- Receptor grid generation
- GPU Shape screen
- Docking model optimization
End of module checkpoint
Module 3
5 Hours + compute time
Post-screening hit evaluation and prioritization
Videos
- Clustering and scoring methods
- Chemical diversity & library design
- Active Learning Glide
- Glide WS
Tutorials
- DISE-like selection
- Rescoring and rank comparisons of SBVS and LBVS hits
- Assess AL Glide results: Hit Analyzer
- Running Glide WS on final compounds
End of module checkpoint
Module 4
4 Hours
Scaling workflows using machine learning and FEP+
Videos
- Methods for further evaluating and enriching hits
- FEP+ methods
- Active learning ABFEP+
- Machine learning property predictions
Tutorials
- Evaluating AL-ABFEP+ results
- Using ML predicted properties
End of module checkpoint
Module 5
4 Hours + compute time
Final case study
Video
- Case study overview
- Case study findings and course closing
Tutorial
Virtual screen analysis

Assignment
Absolute binding FEP+ evaluation of chosen hits
Course completion and certification
Need help obtaining funding for a Schrödinger Online Course?
We proudly support the next generation of scientists and are committed to providing opportunities to those with limited resources. Learn about your funding options for our online certification courses as a student, post-doc, or industry scientist and enroll today!
Show off your newly acquired skills with a course badge and certificate
When you complete a course with us in molecular modeling and are ready to share what you learned with your colleagues and employers, you can share your certificate and badge on your LinkedIn profile.
Frequently asked questions
How much does the Virtual screening with integrated physics and machine learning online course cost?
Pricing varies by each course and by the participant type. For students wishing to take this, we offer a student price of $240, and $645 for non-students.
What time are the lectures?
Once the course session begins, all lectures are asynchronous and you can view the self-paced videos, tutorials, and assignments at your convenience. When registering for the course you will select the start and end date. Within those dates, you will have asynchronous access to the course material and virtual workstation to work on the course when it best suits your schedule.
How could I pay for this course?
Interested participants can pay for the course by completing their registration and using the credit card portal for an instant sign up. Please note that a credit card is required as we do not accept debit cards. Additionally, we can provide a purchase order upon request, please email online-learning@schrodinger.com if you are interested in this option. If you have any questions regarding how to pay for the course, please visit our funding options page.
How can I preview the course before registering?
Are there any scholarship opportunities available for students?
Schrödinger is committed to supporting students with limited resources. Schrödinger’s mission is to improve human health and quality of life by transforming the way therapeutics and materials are discovered. Schrödinger proudly supports the next generation of scientists. We have created a scholarship program that is open to full-time students or post-docs to students who can demonstrate financial need, and have a statement of support from the academic advisor. Please complete the application form if you qualify for our scholarship program!
Will material still be available after a course ends?
While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course.
Do I need access to the software to be able to do the course? Do I have to purchase the software separately?
For the duration of the course, you will have access to a web-based version of Maestro, Bioluminate, Materials Science Maestro and/or LiveDesign (depending on the course). You do not have to separately purchase access to any software. While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course. Please note that Schrödinger software is only to be used for course-related purposes.
What courses are recommended to take in conjunction with this online course?
The Target enablement, preparation, & validation & Designing quality ligand libraries online courses are recommended to be taken before, during, or after the virtual screening with integrated physics and machine learning online course. These offerings are all closely related and are intended for similar audiences. You will notice overlap of some foundational theory videos as these each contain key concepts relevant to each facet of the virtual screening pipeline. However, each course is uniquely focused and is able to elaborate on specific guidance related to the course topic.
Related courses
 Life Science
Life Science
Life Science
Life Science
Designing quality ligand libraries
Exploring chemical space, profiling and tailoring ligand libraries, validating docking models, and methods of enumeration for hit discovery
 Life Science
Life Science
Life Science
Life Science
Target enablement, preparation, & validation
Enabling protein structures from x-ray crystallography, cryo-EM, ML-methods, and homology modeling for structure-based computational workflows
 Life Science
Life Science
Life Science
Life Science
Free energy calculations for drug design with FEP+
Running, analyzing, and troubleshooting relative binding FEP+ calculations for small molecule lead optimization
Efficient long-range machine learning force fields for liquid and materials properties
Applied Machine Learning for Formulations
Optimization of Formulations Using Machine Learning
Machine Learning for OLED Device Design
Advancing drug discovery programs with machine learning-enhanced de novo design


MAY 21, 2025
Advancing drug discovery programs with machine learning-enhanced de novo design
De novo molecular design creates entirely new chemical entities from scratch, accelerating drug discovery by generating billions of novel molecular structures. Subsequent computational profiling of these ideas harnesses physics-based calculations and machine learning algorithms to rigorously and rapidly predict experimental endpoints for this vast chemical space.
In this webinar, we will demonstrate how large-scale de novo design workflows in Schrödinger’s AutoDesigner, combined with rigorous free energy-based scoring methods, have been applied to several recent programs to overcome critical design challenges. We will outline the use of de novo design with AutoDesigner to accelerate an EGFR discovery project, enabling the exploration of 23 billion novel chemical structures and identifying four novel scaffolds with favorable potency and property profiles in just six days. We further highlight de novo core design strategies applied to WEE1 inhibitor development, in which an automated approach generated entirely new chemotypes achieving >10,000X selectivity over PLK1 while maintaining potent target inhibition.
Finally, we introduce AutoDesigner LinkerDesign, a workflow capable of de novo generation and evaluation of billions of potential linkers between molecular fragments, further expanding computational design capabilities. We conclude with an overview of how we track the impact of these tools using interactive dashboards in LiveDesign.
Webinar Highlights
- How to design novel cores for hit identification and R-groups and linkers for hit-to-lead and lead optimization using AutoDesigner
- Examples of dramatically improving potency and selectivity in several drug discovery programs
- Requirements and best practices to apply the technology to your drug discovery programs
- Methods for tracking key performance metrics using dashboards in LiveDesign
Our Speakers

Pieter Bos
Principal Scientist II, Schrödinger
Pieter Bos, Ph.D., is a principal scientist and product manager of AutoDesigner and De Novo Design workflows. At Schrödinger, his main focus is the research, development and optimization of automated compound design algorithms. Lead scientist for the design and execution of enumerated drug molecule libraries for internal and collaborative drug design projects. He received his Ph.D. in Synthetic Organic Chemistry from the University of Groningen in the laboratory of Prof. Ben Feringa. Prior to joining Schrödinger, he worked as a postdoctoral researcher in synthetic methodology development at Boston University (Prof. John Porco and Prof. Corey Stephenson) and small molecule drug discovery at Columbia University (Prof. Brent Stockwell).

Sathesh Bhat
Executive Director, Therapeutics Group, Schrödinger
Sathesh Bhat, Ph.D., executive director in the therapeutics group, joined Schrödinger in 2011. He is responsible for overseeing computational chemistry efforts on internal and partnered drug discovery programs at Schrödinger. Previously, Sathesh worked at both Merck and Eli Lilly leading computational efforts in several drug discovery programs. He obtained his Ph.D. from McGill University, which involved developing structure-based methods to predict binding free energies. Sathesh has co-authored multiple patents and publications and continues to publish on a wide variety of topics in computational chemistry.
Accelerating pharmaceutical formulations using machine learning approaches


APRIL 8, 2025
Accelerating pharmaceutical formulations using machine learning approaches
Machine learning (ML) is revolutionizing pharmaceutical formulation design by enabling data-driven predictions of critical properties such as solubility, viscosity, and stability. Chemistry-informed AI/ML models provide a powerful framework for accelerating materials innovation beyond active pharmaceutical ingredients (APIs) to complex drug formulations. The ability of machine learning to analyze large amounts of data and make predictions about new formulations allows for rapid exploration of vast chemical spaces, significantly reducing the need for traditional trial-and-error experimentation. Automated workflows can integrate chemical composition and molecular structure to generate predictive models, optimizing formulation properties with greater speed and efficiency.
In this webinar, we will demonstrate how Schrödinger’s integrated ML- and physics-based approaches are transforming pharmaceutical formulation design, including:
- How an automated ML workflow, incorporating chemistry and composition, can predict API solubility in binary solvents
- How ML models, augmented with physics-based descriptors, can be used to optimize viscosity predictions of organic molecules for better formulation performance
- How formulation ML tools empower non-experts to design novel drug formulations that satisfy multiple target criteria simultaneously
Our Speakers

Anand Chandrasekaran
Senior Principal Scientist, Schrödinger
Anand Chandrasekaran joined Schrödinger in 2019 and currently serves as the Product Manager for MS Informatics. His expertise lies in applying machine learning across various domains within materials science and computational modeling. He earned his Ph.D. in Materials Science under Prof. Nicola Marzari at the Swiss Federal Institute of Technology, Lausanne. Prior to joining Schrödinger, Anand worked with Prof. Rampi Ramprasad, focusing on polymer informatics, machine learning force fields, and machine learning for electronic structure calculations.

Shiva Sekharan
Senior Director, Schrödinger
Shiva Sekharan, Ph.D., is the Senior Director of Formulations Business Development at Schrödinger and is responsible for driving the business development efforts in the formulations space. Shiva is an experienced business development executive in the CRO and AI-based services and software solutions industry and has several years of experience in managing business accounts, customer relationships, and expectations with clients in the pharmaceutical, agrichemical, and academic industries across the US, Europe, and Asia territories. His expertise lies in identifying new business opportunities among existing customers, devising sales and collaboration strategies for customer expansion,and ensuring top-tier services, products, and knowledge-driven solutions are available 24/7 to customers across the globe. Before joining Schrödinger in 2023, Shiva held a BD role at XtalPi Inc., where he led the US solid-state services unit, worked with departmental heads to establish effective goals, sales targets, outline procedures and best practices, and provide strategic directions to increase revenue. Shiva earned his Ph.D. in Theoretical Chemistry from the University of Duisburg-Essen, Germany, followed by postdoctoral stints at the Max-Planck Institute for Polymer Science, Emory University, Fukui Institute for Fundamental Chemistry, and Yale University. Shiva is an accomplished computational chemist, with strong research expertise in the areas of quantum chemistry and drug discovery (>40 publications, >1100 citations, H-index = 20).
What our alumni say