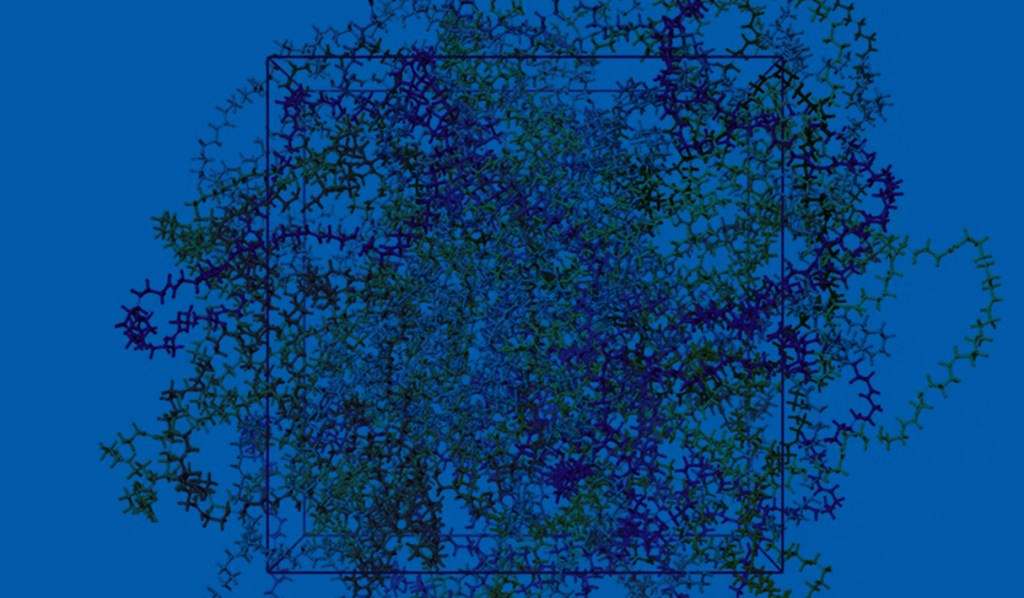
Machine learning force fields (MLFFs), also known as machine learning interatomic potentials, represent an intermediate between classical force fields and density functional theory (DFT), maintaining the linear scaling of the former while approaching the accuracy of the latter. Beyond the balance of accuracy and efficiency/cost, MLFFs are enabling new scientific insights by making large-scale and longtime scale simulations feasible for reactive systems. This opens the door to modeling complex materials systems that were previously computationally prohibitive with traditional quantum methods.
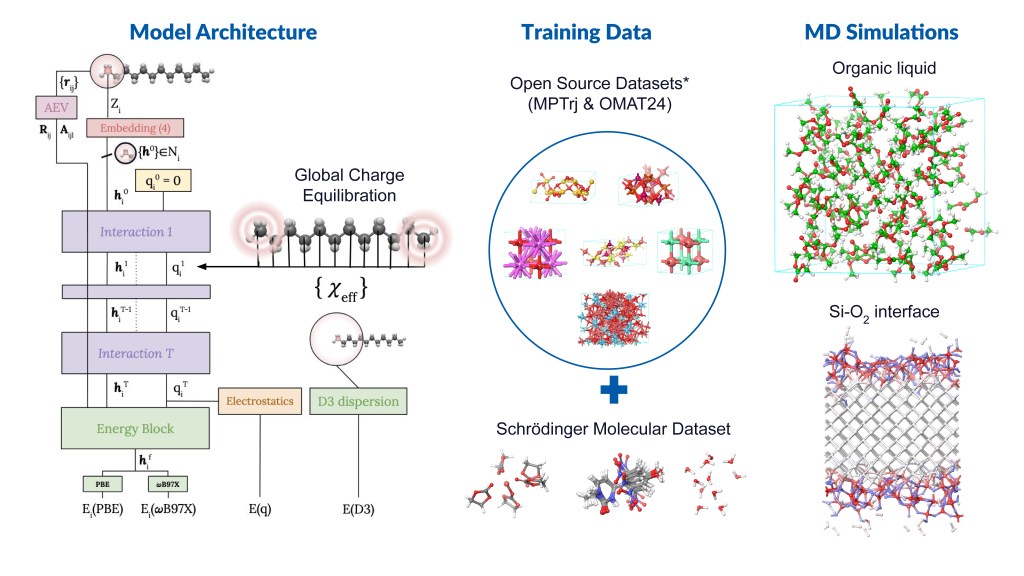
Message Passing Network with Iterative Charge Equilibration (MPNICE) is an MLFF architecture developed by Schrödinger for which multiple pretrained models spanning 89 elements are available, and which explicitly incorporates equilibrated atomic charges and long range electrostatics.1 This technological advancement has removed the drawback of previous MLFFs that were limited by the number of unique atomic elements they could model. Furthermore, inclusion of atomic charges and electrostatics through charge equilibration has enabled representation of multiple charge states, ionic systems, and electronic response properties, while simultaneously improving accuracy. In addition to MPNICE, the Schrӧdinger suite also allows users to utilize the Universal Models for Atoms (UMA),2 developed at Meta. This suite of models offers very high accuracy, includes a model that yields good performance for reaction barrier heights for finite systems, and covers the majority of the periodic table.
By integrating state-of-the-art MLFF methods with high-performance OPLS4 or OPLS5 force fields, as well as advanced DFT and molecular dynamics (MD) engines, Schrödinger offers a uniquely powerful platform for materials simulation — positioning us as the leading partner in advanced MLFF technologies. In this application note, we present case studies from materials-intensive industries, including batteries and catalysis.
Benefits of MLFF
Near DFT-level accuracy with orders of magnitude reduction in computational time
Option for GPU accelerated molecular dynamics with Desmond
Large chemical space spanning 89 elements
Specialized force fields for organic, inorganic, and hybrid materials
Figure 1: Illustrative examples of Schrödinger’s MLFF workflow and its applications *Datasets references: Nature Machine Intelligence 2023, 5, 1031–1041; arXiv:2410.12771
Diverse applications of MLFF
Batteries:
Calculate bulk and transport properties, such as diffusion, viscosity, and conductivity of liquid electrolytes
Simulate Li-ion diffusion in solid-state electrolytes and cathode coating materials
Model electrolyte reactivity and SEI formation
OLED materials:
Simulate molecular packing and thin-film morphology
Investigate doping, host-guest, and interlayer interactions
Link device properties to the static and dynamic disorder of molecular systems
Facilitate thermomechanical property prediction
Model charge and exciton transport
Crystal structure prediction:
Rank order organic crystal structures
Adsorption on surfaces:
Study reactivity of multiple adsorbates in extended models of complex surfaces
Reactivity in molecules and solid-state:
Investigate reaction pathways and transition states
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Cutting-Edge Cosmetics: Innovating for Sustainability with Machine Learning & Molecular Simulations
Share
Speaker
Jeffrey Sanders
Product Manager, CPG
Abstract
Demand for sustainable, eco-friendly cosmetics is growing, but meeting customer demand requires finding formulations that perform at least as well as the existing synthetic alternatives. In this webinar, we’ll explore the challenges chemists face, and how new approaches can help find solutions quicker.
Harnessing advances in machine learning, particularly active learning combined with molecular simulation, holds immense potential for efficient formulation development in the eco-friendly cosmetics industry. However, the limited availability of relevant data poses challenges. Active learning bridges this gap by integrating diverse datasets, enabling the construction of robust machine learning models that cover the extensive design space. Molecular simulation complements this process by predicting physical properties of various formulations.
In this hour-long, interactive webinar, you will hear about the efficacy of this approach using rhamnolipid biosurfactants as an eco-friendly formulation example.
By attending this webinar you will learn:
New digital approaches to sustainable cosmetic formulation development
How machine learning methods and molecular simulations reduce formulation development cycle time
To identify key areas in your R&D where machine learning and molecular simulations can provide value
Tremendous demands for development of new materials with improved performance and greener chemistry
Pressure to bring products to market quickly and cost-efficiently
Rapid need for informatics-based predictive models to extend the scale of materials optimization and discovery
Limited availability of large datasets of materials properties to effectively train models
Solutions
Amplify physics-based modeling with advanced machine learning technology and deep learning capabilities
Leverage machine learning to extend predictions over extensive chemical space unavailable to experimental methods
Develop customized descriptors for highly diverse material systems to improve the accuracy of predictions
Collaborate, manage disparate data, and share predictive models across project teams with a unified cloud-based enterprise informatics platform
With the rapid development of technology in the fields of energy, aerospace, electronics, foods and more, there are increasing demands for the design and discovery of new materials. To meet these demands, materials scientists seek innovative methods to optimize chemical properties and reduce time-to-market of better performing, more sustainable products. However, relying solely on traditional trialand- error experimental methods has become too costly and too slow, and is limited by experimental conditions. Thus the materials innovation R&D cycle has long benefited from the use of physics-based simulation engines such as quantum mechanics and molecular dynamics to help lower the cost of discovering novel chemistries, structures, morphologies, and compositions of materials for a wide array of applications and industries. In the past few years, the growth of computational power and the interest in building large datasets of materials properties has led to the growing adoption of materials informatics and machine learning-powered approaches in materials science. However, such methods are highly data intensive and suffer from an inability to extrapolate beyond the chemical space of the training model. There is a pressing need for rapid, informatics-based predictive models to extend the scale of materials optimization and discovery, extract property limits and design rules, and drive the natural synergy between physics-based modeling and machine learning methods. Additionally, with the rapid advance of machine learning methods, there is an increasing need to deploy and share accurate predictive models across research organizations for use by experts and non-experts alike. Successful R&D digitization efforts require a robust, collaborative platform for assessing predictive models for use in research.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Advancing a Pipeline of Drug Discovery Programs Using Diverse Machine Learning Strategies Coupled with Physics-Based Simulations
Share
Speakers
Jennifer Knight, Director at Schrödinger
Karl Leswing, Executive Director, Machine Learning at Schrödinger
Abstract
The application of machine learning (ML) to drug discovery is growing rapidly, ranging from target discovery to molecular design and patient stratification. However, the lack of large, high quality datasets on which to train models limits their current utility, particularly in early discovery. Complementing ML strategies with physics-based approaches offers a way to overcome these limitations and expand the domain of applicability of ML models.
In this presentation, we describe how we are integrating machine learning strategies with physics-based modeling technologies within Schrödinger’s Therapeutics Group – from hit discovery to lead optimization and drug formulation – and the impact of these approaches on active drug discovery programs.
Key Takeaways:
Active learning strategies enable routine, efficient exploration of target-specific chemical space as well as protocol optimization to streamline project work and decisions
Automated machine learning engines within LiveDesign allow whole teams to iterate through training, reviewing and deploying models to drive the drug design and optimization process
Deep neural networks trained on QM DFT calculations (QRNN) can help guide in silico crystal polymorph prediction for de-risking drug formulation
Generative machine learning approaches can design into new, highly-targeted chemical space
Accelerating catalysis and reactivity r&d with atomic-scale simulation, machine learning, and enterprise informatics
As market demands evolve, R&D scientists across industries — from consumer packaged goods to automotives — face similar challenges in developing and optimizing the next-generation of catalysts. Scientists need new catalysts that help them reduce energy requirements, eliminate unwanted side products, and improve the selectivity and reactivity of reactions.
We need new materials, and we need them fast. Fortunately, the continual march of scientific progress promises faster answers than in the past. Computer-driven molecular design, with its ability to generate massive quantities of simulated data, facilitates entry into new frontiers of chemical discovery for sustainable materials design. It brings the promise of speed and accuracy, and it allows R&D scientists to scan through large molecular space to triage down and experimentally test only the most promising chemistries.
Schrödinger’s Materials Science platform leverages the power of physics-based simulation, machine learning, and enterprise informatics to enable the optimization and discovery of effective and selective catalysts and reactive systems by offering:
Differentiated model builders
Interactive visualization and analysis tools
Highly-efficient density functional theory (DFT) engines
Customizable automated reaction workflows
A collaborative enterprise platform solution
Application Overview
Catalysts
Advance the knowledge of catalytic mechanisms: Simulate catalytic pathways to fundamentally understand the forces providing reactivity and selectivity
Improve the lifetime of catalysts: Predict stability and degradation of catalysts and products
Design and optimize catalysts: Automatically run high-throughput screening of catalysts by leveraging known mechanistic pathways with novel catalysts
Benefit from inherently chemically agnostic workflows: Accelerate and automate any reaction mechanism in both homogeneous and heterogeneous screening workflows
Reactive Systems
Understand reaction mechanism and pathways: Simulate reaction thermodynamics, kinetics, reaction rates, and barriers
Accelerate product development: Automatically predict selectivity and activity of reactions with a library of interesting substrates and catalysts
Optimize product properties: Compute tacticity of polymers with relative reaction pathway energies
Predict product distribution: Automatically locate transition state
Drive innovation in reaction design: Study effect of steric, electronic, and ligand on the reactivity
Team Collaboration and Digital Data Management
Empower team collaboration: Employ webbased enterprise informatics tools for sharing experimental and predictive models seamlessly
Amplify research with improved decisionmaking: Rapidly deploy automated reaction workflows and machine learning models to drive large-scale predictions and assist novel design approaches
Improve project management: Accelerate project communication and collective learning by capturing, analyzing, and testing new ideas and data in a centralized platform
Products
Discover the Schrödinger products that enable your success in the catalysis industry.
Selected publications
Iron-catalysed Synthesis and Chemical Recycling of Telechelic 1,3-enchained Oligocyclobutanes.
Chirik P. J. et al. Nature Chemistry 2021, 13, 1 56-162.
Exploring the Mechanism of Cr(VI) Catalyzed Hypochlorous Acid Decomposition.
Busch M et al. ChemCatChem 2022, e202101850.
Intramolecular Hydroxyl Nucleophilic Attack Pathway by a Polymeric Water Oxidation Catalyst with Single Cobalt Sites.
Sun L. et al. Nature Catalysis 2022, 5, 414–429.
Olefin Metathesis Catalyzed by a Hoveyda– Grubbs-like Complex Chelated to Bis(2- mercaptoimidazolyl) Methane: A Predictive DFT Study.
Martínez J. P. et al. J. Phys. Chem. A 2022, 126, 5, 720–732.
Mechanistic Study of Metal–Ligand Cooperativity in Mn(II)-Catalyzed Hydroborations: Hemilabile SNS Ligand Enables Metal Hydride-Free Reaction Pathway.
Baker R. T. et al. ACS Catal. 2021, 11, 15, 9043–9051.
Cut-off Scale and Complex Formation in Density Functional Theory Computations of Epoxy-Amine Reactivity.
Laurikainen P. V. et al. ACS Omega 2021, 6, 44, 29424–29431.
Decarbonylative Fluoroalkylation at Palladium(II): From Fundamental Organometallic Studies to Catalysis.
Sanford M. S. et al. J. Am. Chem. Soc. 2021, 143, 44, 18617–18625.
One-Pot Chemo-bioprocess of PET Depolymerization and Recycling Enabled by a Biocompatible Catalyst, Betaine.
Kim K. H. et al. ACS Catal. 2021, 11, 7, 3996–4008.
Automated Transition State Search and Its Application to Diverse Types of Organic Reactions.
Friesner R. A. et al. J. Chem.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
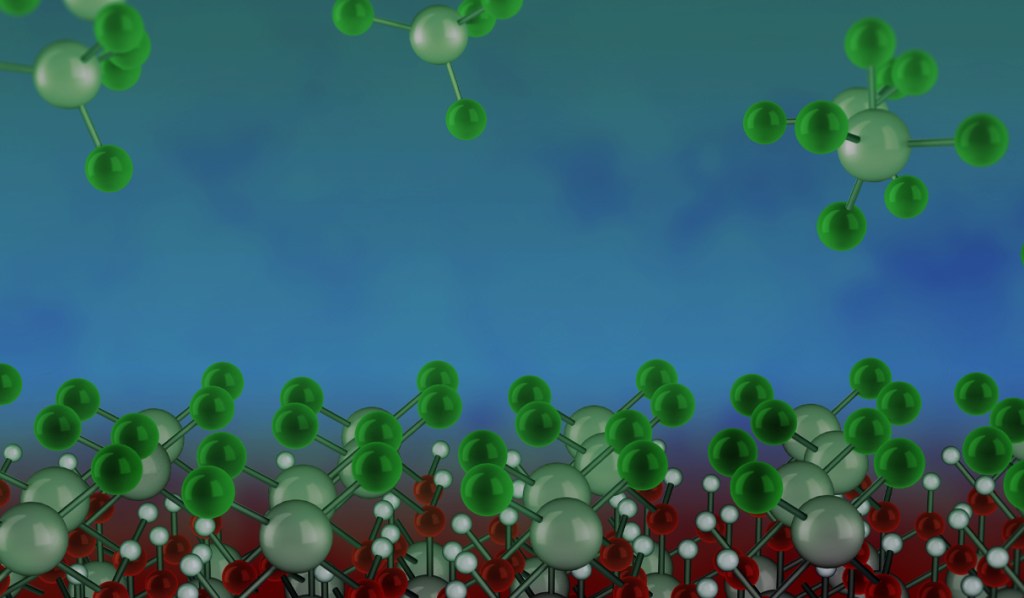
The Schrödinger Materials Science Suite offers computational tools specifically tailored to studying the gas-surface chemistries of atomic layer deposition and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (usually with density functional theory, DFT) and prediction of key properties. This modeling approach thus has a unique role to play both in deepening our understanding of existing processes and in discovering novel chemicals.
Many of today’s high-tech devices are manufactured by processing materials at the nanoscale, including computer chips, data storage and communications devices, sensors, solar cells and batteries. Making devices smaller, more powerful and more energy-efficient means developing new patterning, deposition and etch techniques at ever-finer resolution, in some cases down to just a few atoms thick. Chemical processing is being pushed to its limits in terms of purity, uniformity, conformality and substrate-selectivity.
Optimizing a deposition or etch process to more stringent tolerances requires a deep understanding of the underlying chemistry. Integrating new materials into a device often requires the discovery of new chemicals with improved properties. In both respects, researchers are increasingly turning to computer simulations to cut down on lab work and make discoveries faster.
The Schrödinger Materials Science Platform offers computational tools specifically tailored to tackling this problem, by studying the gas-surface chemistries of deposition, etch and related nanofabrication processes. Schrödinger software is designed for rapid and automated enumeration of chemical space, detailed study of gas-surface chemistries at the quantum mechanical level (with density functional theory, DFT) and prediction of key properties. This modeling approach can both deepen our understanding of existing processes and help discover novel chemicals.
Automate Precursor Screening for Improved Properties
Organometallic complexes can be used to deposit or etch pure metals or compounds such as oxides or nitrides. However, synthesis of the complexes in the lab can be time-consuming and difficult because many complexes react violently in air. Subsequent characterization and testing in a reactor is even more costly. Digital chemistry can rapidly screen larger numbers of chemicals than can ever be experimentally tested, narrowing down the chemical space and discovering the most promising candidate molecules for deposition or etch.
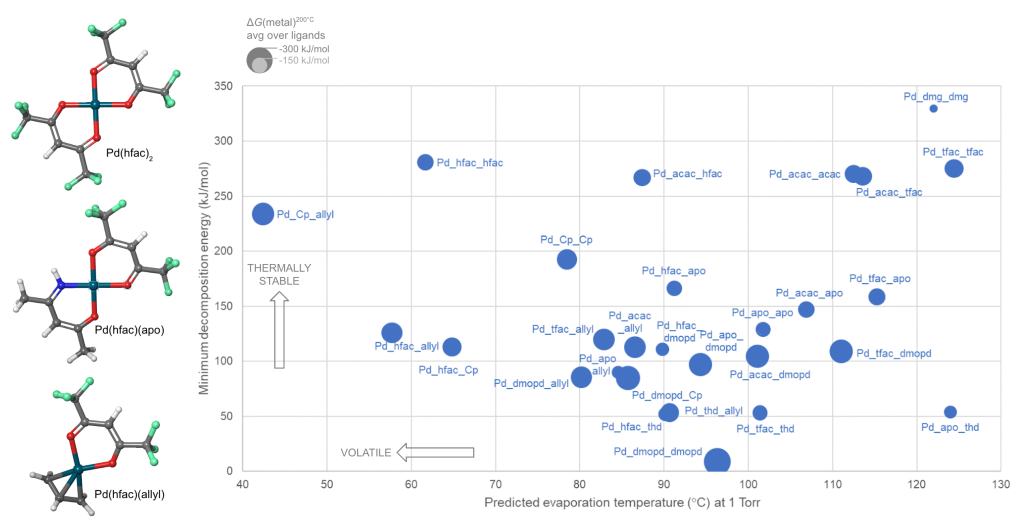
Schrödinger provides easy-to-use software for automated generation and assessment of hundreds or thousands of precursor gases. A library of hundreds of commonly-used ligands is provided, including bidentate and haptic ligands, and the library can be customized. A selection from the library is then used to enumerate and build all possible precursor complexes. We present here a case study in which a library of 10 ligands form 55 distinct complexes with divalent palladium (Pd) as the metal cation and some of these candidate complexes are shown in Figure 1.
Synthesizability
Combining two or more different ligands in a heteroleptic complex is a promising way to tune precursor properties, but it is difficult to guess whether a given combination can in fact be synthesized. This question can be answered by computing the energy for ligand exchange back to homoleptic complexes. In the Pd example, this reveals that 14 of the heteroleptic complexes are unstable and can not be synthesized.
Reactivity
Precursors for deposition must be carefully designed so as to react cleanly and deposit the desired solid material, via thermolysis or plasma activation (chemical vapor deposition, CVD) or via reactions with adsorbed fragments (atomic layer deposition, ALD). Likewise, reagents for etching must react with surface atoms to form gaseous products. Lists of candidate precursor molecules can be efficiently screened against deposition or etch chemistries using Jaguar, Schrödinger’s code for molecular DFT. Figure 1 displays the computed reactivity of the complexes with respect to Pd deposition.
Volatility and Thermal Stability
To be useful in a real CVD or ALD process, a precursor must also be volatile and thermally stable during storage and delivery. This limits the process to a window of viable temperatures, with insufficient vapor pressure at lower temperatures and impurities from decomposition at higher temperatures. For instance, Pd(thd)2 suffers from decomposition when heated to 200-250°C for the ALD of Pd metal.1, 2 Calculating volatility and thermal stability with Schrödinger’s tools gives information on how to tune these properties and widen the temperature window for maximum process flexibility.
Schrödinger has used machine learning (ML) to develop a unique model for predicting the evaporation or sublimation temperature of organometallic complexes at a given pressure. The ML-volatility model is accurate to an average of ±9°C over the training set and can evaluate hundreds of molecules per second.
The thermal stability of precursor gases can be quantified by considering the unimolecular decomposition channels: bond dissociation and β-H elimination. All such reactions in each molecule are automatically computed at the DFT level and the lowest energy reaction is selected as the most likely. The minimum decomposition energy of each complex is thus a measure of its thermal stability.
These approaches were applied to compute the properties of the Pd complexes, and Figure 1 shows how this allows the candidate precursors to be assessed.
Rational innovation is facilitated by Schrödinger’s atomistic modeling tools. Novelty is maximized by using a large library of ligands to build a wide variety of complexes, homoleptic and heteroleptic. Risk is minimized by selecting for lab study only those precursors computed to properly combine the desired properties of synthesizability, volatility, reactivity and stability.
Figure 1: Screening novel divalent Pd precursors for deposition. Three sample precursor structures are shown on the left. The x-axis shows ML-predicted volatility and the y-axis shows thermal stability. The area of the circles corresponds to the DFT-computed reactivity towards deposition of Pd metal. The most volatile and stable complexes are towards the top left of the graph.
Gain Insights Into Competing Processes for Optimum Deposition Conditions
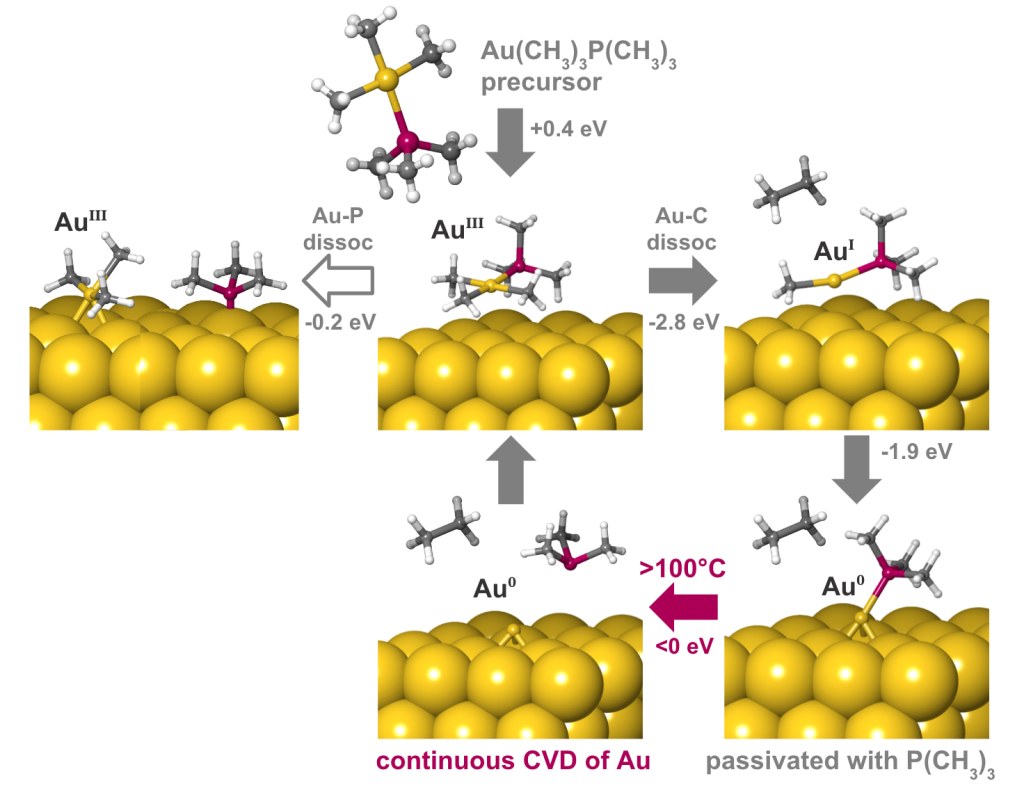
A deposition or etch process consists of a sequence of individual reaction steps, often in competition with other reactions. Simulations give atomic-level insight into these complicated mechanisms and how they depend on reactor conditions such as temperature and pressure. Here we investigate the mechanism of gold deposition from the single-source precursor trimethylphosphinotrimethylgold(III). Experiments show that gold films grow in a self-limiting fashion when this precursor is pulsed at low temperature, switching over to continuous CVD at 100°C and higher, but the reason is unknown.3
To explain this behavior, the Quantum ESPRESSO code for periodic DFT has been used to examine the detailed mechanism of reactive adsorption of the precursor onto a metallic gold surface. Thermodynamics of competing reaction steps have been computed as a function of process temperature and pressure with Schrödinger’s tools for adsorption and surface chemistry [4].
The results are summarized in Figure 2. The physisorbed precursor is in a metastable state, from which dissociation of Au-P or Au-C bonds can lead to chemisorption. Au-C dissociation and elimination of C2H6 is found to be the favored pathway, due to a strong thermodynamic driving force towards reduction of Au(III) to Au(I) and eventually Au(0). Trimethylphosphine is computed to bind to the surface, forming an adlayer that passivates against further precursor adsorption. This explains the low rate of self-limited deposition at <100°C. The computed free energies also reveal that desorption of trimethylphosphine becomes favored at higher temperatures, in agreement with the crossover to CVD seen experimentally at >100°C.
Finding out which bond breaks and how the surface is passivated opens up chemically-informed strategies for improving the process, for example by choosing a suitable co-reagent or modifying the precursor structure. Digital chemistry allows rational design to take the place of trial-and-error.
This example illustrates how changes in process behavior can be traced back to competing chemical pathways. DFT simulations give unique qualitative insight into atomic structure, bonding and oxidation states of the intermediates along each reaction pathway. In addition, quantitative information about kinetics and thermodynamics as a function of process temperature can be computed, giving valuable guidance on how to optimize the nanofabrication process. The result is improved control of purity, uniformity, phase and substrate-selectivity.
Figure 2: Mechanism of deposition of gold from trimethylphosphinotrimethylgold(III) computed with DFT, including free energies evaluated at 100°C.
Conclusion
These cases illustrate how Schrödinger’s Materials Science Platform provides powerful and customizable computational solutions to accurately model surface reactivity, predict precursor properties and discover novel chemicals for optimized materials deposition or etch processes.
References
Low temperature atomic layer deposition of noble metals using ozone and molecular hydrogen as reactants,
J. Hämäläinen et al., Thin Solid Films 2013, 531, 243-250, DOI 10.1016/j.tsf.2013.01.091.
Atomic layer deposition of noble metals: Exploration of the low limit of the deposition temperature,
T. Aaltonen et al., J. Mater. Res. 2004, 19, 3353–3358, DOI 10.1557/JMR.2004.0426.
Reaction mechanism of the Me3AuPMe3–H2 plasma-enhanced ALD process, M. Van Daele et al.,
Phys. Chem. Chem. Phys. 2020, 22, 11903-11914, DOI 10.1039/C9CP06855D.
First-principles investigation of crossover between ALD and CVD in the thin film deposition of gold,
C. N. Brock. D. J. Giesen, A. Fonari & S. D. Elliott, Materials Research Society Spring Meeting, 2022.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Driving innovation in polymer R&D with molecular simulation & machine learning
R&D scientists across broad industries from aerospace to electronics face challenges in developing the next-generation of polymers and composites with high-performance, multifunctional capabilities that also meet society’s demands for miniaturization and environmental sustainability.
Chemical reactivity, physical morphology, and polymer physics drive the behavior of polymers and soft materials. Schrödinger’s Materials Science platform provides a unique combination of capabilities for the design and optimization of polymers, including:
Automated thermophysical and mechanical response workflows
Chemically adaptable cross-linking workflows
Analysis tools for the simulation, optimization, and discovery of novel polymers including linear, crosslinked, elastomers, dendrimers, and copolymers
Schrödinger’s tailored solutions for molecular modeling of polymers, mixtures, and composites can reduce cost and risk, shorten timelines and drive innovation in a broad range of industries.
Industries and Applications
Automotive and Aerospace
Optimize polymer formulation properties: Model water transport, uptake and morphological stability in polymer composites using multistate simulations
Accelerate chemical design to properties critical to product and processing constraints: Identify the unique combinations of epoxy-amine reactions for target properties using highthroughput screening
Accelerate the manufacturing process pipeline: Predict polymer gelling during manufacturing process
Design greener products that are more environmentally sustainable: Simulate and predict properties of high-performance resins with bio-based materials, and automate discovery of new biomaterials
CPG Packaging and Formulation
Develop environmentally sustainable packaging: Study interfacial interactions between packaging materials and consumer goods, and simulate water uptake for barrier design and performance
Optimize polymer formulation: Quantify the diffusion of additives in polymer packaging materials
Minimize production waste: Understand the potential causes and impact of polymer production deviations
Innovate with natural materials: Explore active, recyclable, and bio-based materials through molecular simulation
Reduce processing cost: Evaluate new formulations for potential manufacturing challenges
Specialty Polymers
Speed up decision-making on catalyst selection for raw materials production: Simulate and understand the catalysis mechanisms, selectivity, and reactivity of epoxy amine, urethane, and other reactions
Optimize the design of high-performance polymers: Predict glass transition, thermal stability, and thermal expansion with new polymers
Drive innovation in polymer reactions: Predict curing kinetics and processing properties
Electronic Packaging
Identify thermal/mechanical performance issues early in design: Simulate coefficient of thermal expansion for packaging polymers
Optimize chemical stability: Simulate interactions of packaging polymers with processing solvents and water to predict stability in use
Design new monomers for improved performance: Predict design drivers like Dk, Df and refractive index with physics-based simulations and machine-learned models
Batteries
Understand mechanisms at molecular level: Predict electrolyte degradation reaction mechanisms and control energetics to understand the formation of functional solid electrolyte interphase layers at anodes (SEI) and cathodes (CEI)
Identify best-performing battery materials: Perform high-throughput screening of anode, cathode, electrolyte and additive materials to identify best chemistries for development of nextgeneration batteries
Optimize electrolyte design: Simulate interatomic interactions and transport of ions in liquid and polymer electrolytes
Pharmaceutical Formulation
Optimize drug carrier design: Evaluate the solubility/miscibility of drugs with different polymeric carriers or barriers
Optimize drug formulations: Visualize and characterize pH-dependent polymeric surfactant and drug interactions in solution and precipitation inhibition behavior
Drive effective drug delivery: Simulate drug release profiles from amorphous solid dispersions into solution
Team Collaboration and Digital Data Management
Empower collaboration: Employ web-based enterprise informatics tools for sharing experimental and predictive models seamlessly
Amplify research: Rapidly deploy of machine learning models to drive predictions and assist novel design approaches
Improve project management: Accelerate project communication and collective learning by capturing, analyzing, and testing new ideas and data in a centralized platform
Products
Check on the products that enable your success in the polymer industry.
References
Melt-state degradation mechanism of poly (ether ketone ketone): the role of branching on crystallization and rheological behavior.
Wiggins J. et al. Polym. Degrad. Stab. 2022, 200, 109968.
Atomistic Molecular Dynamics in PEO/PMMA Blends Having Significantly Different Glass Transition Temperatures.
Habasaki J, Int. J. Appl. Glass Sci, doi.org/10.1111/ijag.16553.
Experimental Investigations of AlMg3 Components with Polyurethane and Graphene Oxide Nanosheets Composite Coatings, after Accelerated UV-Aging.
Murariu A. C. et al. Molecules 2022, 27, 1, 84.
Polycyanurates via Molecular Dynamics: In Situ Crosslinking from Di(Cyanate Ester) Resins and Model Validation through Comparison to Experiment.
Moore L. M. J. et al. Macromolecules 2021, 54, 13, 6275–6284.
Photophysical Properties of Cyclometalated Platinum(II) Diphosphine Compounds in the Solid State and in PMMA Films.
Anderson C. M. et al. ACS Omega 2021, 6, 42, 28316–28325 High-Throughput Molecular Dynamics.
Simulations and Validation of Thermophysical Properties of Polymers for Various Applications.
Afzal. A et al. ACS Appl. Polym. Mater. 2021, 3, 2, 620–630 Fundamental Limits to the Electrochemical Impedance Stability of Dielectric.
Elastomers in Bioelectronics.
Floch P. L. et al. Nano Lett. 2020, 20, 1, 224–233.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
A crucial process in manufacturing CPUs and other high-tech devices is the deposition of solid material from reactive vapors. Different precursor vapors are used for chemical vapor deposition, vapor phase epitaxy, atomic layer deposition – and indeed the reverse process of atomic layer etching – with the precursor chemistry carefully designed for each case so as to control material quality at the nanoscale. But what all these techniques have in common is that the precursor chemicals must evaporate or sublime at a low enough temperature. Too much heating when vaporizing a precursor can make it decompose, causing it to be undeliverable to the growing surface.
With volatility playing such a central role in this technology (and in other fields like distillation, refrigeration, inkjet printing, food and perfumes), it is surprising that we understand so little about it. Volatility is the product of a remarkably fine balance of interatomic forces, dictating the extent to which molecules condense together as a solid or liquid, or bounce apart into a vapor and deliver a certain vapor pressure at any given temperature. These interatomic forces can be computed very precisely with quantum mechanics for one molecule or a group of molecules, but not at the scale of a liquid or solid. Even with today’s computing power, routinely and accurately predicting precursor volatility ‘from first principles’ remains unfortunately out of reach.
Machine learning approaches
Could an alternative more empirical approach prove useful? Does enough experimental data exist to find the relation between volatility and chemical structure? The vaporization of some organic molecules, such as alcoholic fractions or natural fragrances, has been of interest for centuries and high-quality vapor pressure data are available in the literature. Over the last decade, these data have been analyzed with advanced fitting algorithms that come under the umbrella of ‘machine learning’. Schrödinger has leveraged the latest machine learning techniques to develop a highly-accurate model that predicts the volatility of organic molecules up to C20.
However, when building machine learning models to predict volatility of precursor molecules, which are typically organometallic complexes, the situation is not so straightforward. New precursor molecules are constantly being proposed and evaluated. Commercial sensitivity sometimes means that data are partially withheld, or plagued by experimental configuration differences from laboratory to laboratory. Additionally, for the common aim of material processing, complete pressure-temperature curves are rarely measured, as it is more pragmatic to focus on the temperature for vapor to transport successfully to the reactor. As a result, datasets for building predictive models are sparse and incomplete.
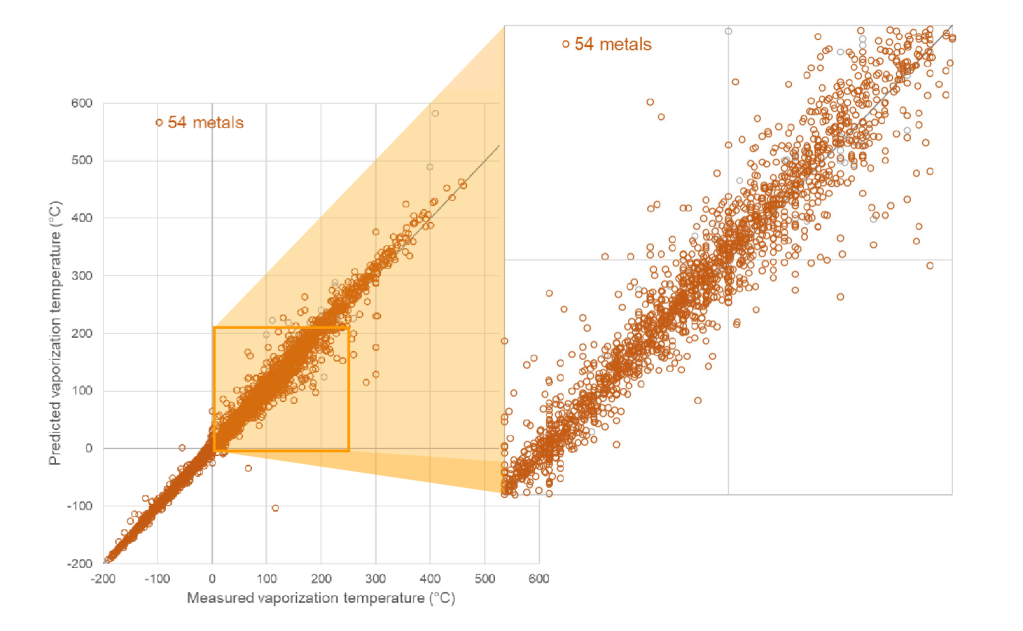
Prediction of volatility for inorganic and organometallic complexes
Schrödinger scientists embarked on the challenge of building machine learning models to predict the volatility of precursor molecules. Using in-house expertise in machine learning and advanced informatics, Schrödinger scientists collated and digitized information about organometallic precursors from disparate literature sources and applied a variety of machine learning algorithms (such as Random Forest and Neural Networks) in conjunction with different chemoinformatic descriptors and fingerprints. The result is the first capability of its kind for accurately and efficiently predicting the volatility for inorganic and organometallic complexes from their chemical structures. For complexes of the fifty most common metals and semimetals, the model predicts the evaporation or sublimation temperature at a given vapor pressure with an average accuracy of ±9°C (which is about 3% of the absolute temperature). As a trained model, the turnaround time is fast with the ability to compute hundreds of complexes per second.
New avenues for precursor development
This predictive model opens a new path for designing novel precursors with improved performance, not only improving their deposition or etch chemistry, but also optimizing the temperature at which they evaporate or sublime and can be delivered as a vapor. This advance will allow a much wider range of structural modifications to be screened computationally than before and will produce candidate precursors for experimental synthesis and testing that are both less risky and more innovative. This volatility model, together with Schrödinger’s quantum mechanics-based workflows for computation of reactivity and decomposition, gives scientists a complete design kit for vapor-phase deposition or etch, delivering a faster pace of research into materials and processes for new technologies.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Limited Experimental Data? No Problem: Machine Learning and Physics in Preclinical Drug Discovery
Share
Speakers
Sathesh Bhat, Steven Jerome, and Karl Leswing Executive Director, Sr. Principal Scientist, Research Leader
Abstract
The rise of machine learning and accurate, physics-based modeling have facilitated breakthroughs in preclinical drug discovery, accelerating discovery of compounds with improved chemical properties at reduced cost relative to traditional methods.
Application of cutting-edge machine learning methods enables accurate exploration of significantly larger regions of chemical space through interpolation. Combined with accurate, extrapolative physics-based methods through active learning, hit discovery from purchasable libraries of billions of compounds becomes cost effective for the first time. Similarly, drastic expansion in chemical space searched in design-make-test-analyze cycles during lead optimization are enabled by physics-informed machine learning, resulting in speed and cost advantages.
This webcast will provide three practical examples of the application of machine learning in active drug discovery programs – one for property prediction, one for hit identification, and one for maintaining or boosting affinity through design-make-test-analyze cycles in lead optimization.
Data Scarcity? No Problem. Combining Machine Learning and Physics-based Simulations for Smart Design of Materials
Share
Speaker
Anand Chandrasekaran
Senior Scientist
Summary
The materials innovation R&D cycle has long benefitted from the use of physics-based simulation engines such as quantum mechanics and molecular dynamics to help lower the cost of discovering novel chemistries, structures, morphologies, and compositions of materials for a wide array of applications and industries. In the past few years, the growth of computational power and the interest in building large datasets of materials properties has led to the growing adoption of materials informatics and AI-powered approaches in materials science. However, such approaches are highly data intensive and suffer from an inability to extrapolate beyond the chemical space of the training model. In this webinar we demonstrate, using a number of case studies, that the tradeoffs between accuracy and computational complexity lead to a natural synergy between physics-based modeling and machine learning methods and showcase the ability to apply these methods successfully even in the absence of large datasets. By combining the latest physics-based and data-driven approaches, decision-making process for the materials design is quickly assessed over the extensive chemical design space. We will demonstrate this idea over a few recent case studies including in organic electronics, aerospace, automotive, and semiconductor industry. Advanced machine learning techniques such as active learning, genetic optimization, and deep neural network will be utilized to showcase how key materials properties like vapor pressure, electronic structures, chemical stability, optical characteristics, and thermomechanical properties can be predicted with little to no user bias.
Moving Beyond Spreadsheets: Rational Design of Materials Using Advanced Informatics and Machine Learning
Share
Speaker
Yuling An
Product Manager
Summary
In this webinar, Schrödinger’s Dr. Yuling An will demonstrate that machine learning, which often ignores the underlying physics, and physics-based modeling, which may require intensive computing resources, can naturally complement each other to create not only predictive models but also new materials with desired properties over an extensive design space. The growing urgency to digitize and make use of existing data, both from experiments and from simulations, through machine learning, also heightens the need of materials informatics platforms, which bring together automated computational workflows with data analysis and collaboration to make materials innovation more efficient and successful. Examples include recent progress in de novo OLED materials design and prediction of molecular volatility using Schrӧdinger’s materials informatics platform, LiveDesign.