Leveraging Multitask Learning to Improve the Transferability of Machine Learned Force Fields
Active Learning Applications

Active Learning Applications
Accelerate discovery with machine learning
Amplify discovery across vast chemical space
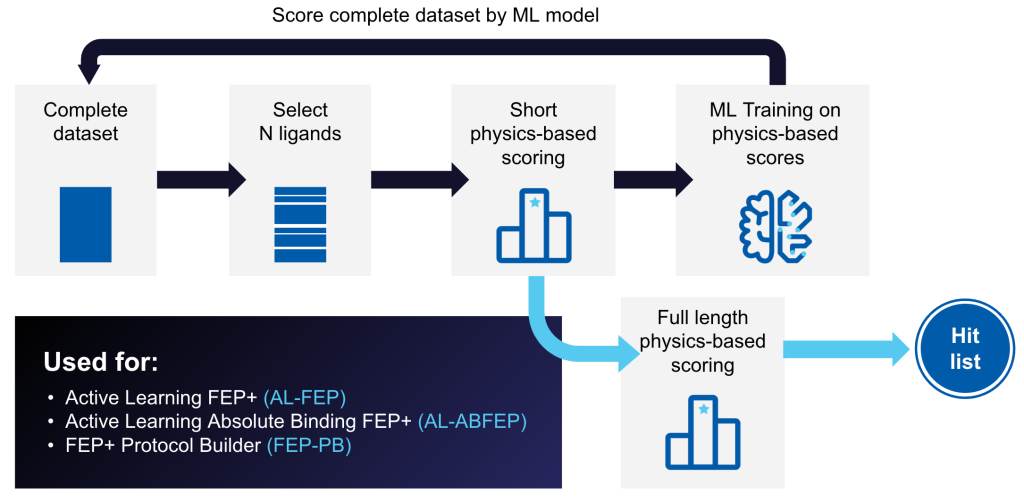
Active Learning Applications is a powerful tool that trains a machine learning (ML) model on physics-based data, such as FEP+ predicted affinities or Glide docking scores, iteratively sampled from a full library.
Trained models can rapidly generate predictions for new molecules and identify the highest-scoring compounds in ultra-large libraries at a fraction of the cost and speed of brute force methods.
Key applications across drug discovery
Active Learning Glide
Find potent hits in ultra-large libraries
Screen billions of compounds with Glide docking amplified by cutting-edge machine learning models in a fraction of the time. Use Active Learning to recover ~70% of the same top-scoring hits that would have been found from exhaustive docking of ultra-large libraries with Glide, for only 0.1% of the cost.
Active Learning FEP+
Explore diverse chemical space in lead optimization
Explore tens of thousands to hundred of thousands of idea compounds with Active Learning FEP+, against multiple hypotheses simultaneously, to quickly identify compounds that maintain or improve potency while achieving other design objectives.
FEP+ Protocol Builder
Expedite FEP+ use for challenging systems with a fully automated workflow
Rapidly generate accurate FEP+ protocols for systems that do not perform well with default settings. FEP+ Protocol Builder uses an Active Learning workflow to iteratively search the protocol parameter space to develop accurate FEP+ protocols, saving researcher time and increases the chances of successfully enabling FEP+.
Learn moreActive Learning Calculator
Glide (Dock All Compounds)
days
compute
cost
cost

Active Learning Glide
Faster
days
compute
cost
cost
For Compounds
Enter in the numbers for your project (type in the box or use slider) to compare compute time and cost.
*Estimated customer compute costs only, based on $0.06 per CPU hour and $.35 per GPU hour. Recommended hardware for AL-Learning Glide.
License costs are not included. Contact us for a quote.
We assume 1M of the best ligands are docked with the final model.
De Novo Design Workflow
Schrödinger’s De Novo Design Workflow is a fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement.
Starting from a hit molecule or lead series, the technology identifies synthetically tractable molecules that meet key project criteria by combining multiple compound enumeration strategies with an advanced filtering cascade (AutoDesigner) and rigorous potency scoring with free energy calculations (Active Learning FEP+).

Featured Webinar
Documentation & Tutorials
Get answers to common questions and learn best practices for using Schrödinger’s software.
Related Products
Learn more about the related computational technologies available to progress your research projects.
De Novo Design Workflow
Fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement

Publications
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Training & Resources
Online certification courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Tutorials
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Other Resources
Accelerating the design and optimization of OLED materials using active learning

Accelerating the design and optimization of OLED materials using active learning
Introduction
OLEDs (Organic Light-Emitting Diodes) are extensively used in digital displays such as those in smart phones, television screens, computer monitors, and game consoles. They contain organic molecules with unique electronic structures that create light in the visible part of the spectrum through a process called electroluminescence. For an OLED to be commercially valuable, it should satisfy several constraints, including high efficiency, low-cost fabrication, and good electrochemical stability for long-term operation, making discovery of novel OLED materials a challenging problem.
Challenges
Molecular modeling and simulation tools have proven effective in materials discovery and are increasingly deployed in industrial R&D. Although digital simulations have offered tremendous time savings for R&D workflows compared to traditional experimental approaches, several challenges persist:
- The potential chemical space for materials design and discovery is massive, even for highly constrained problems
- Predictions based on density functional theory (DFT) calculations can be laborious and computationally expensive, limiting the number of candidates evaluated within fixed timescales and resources
- It is challenging to maintain a high level of accuracy to properly assess the complexity of materials
Solution: Active Learning Workflows
A new approach is required to guide scientists to the best-performing or useful candidates. Thanks to the application of a machine learning (ML) paradigm called “active learning” (AL), Schrödinger has made this problem readily tractable. Recently, Schrödinger has developed active learning workflows which leverage the synergy between physics-based simulations and machine learning for optoelectronic properties predictions. The active learning workflow enables scientists to zoom in on the “best-performing” portion of a given sample space in a more efficient and cost-effective manner. The workflow allows:
- Automated active learning calculations with minimum users input
- Accounting for multiple optoelectronic parameters simultaneously for materials discovery
- Combining active learning with DFT to efficiently identify materials with optimal properties
- Employing built-in descriptors and fingerprints to featurize chemical structures
- Building high-performance ML models using adaptive design procedures
- Minimizing the number of time-consuming physics-based calculations
““The AL workflow enables accelerated design and discovery of optoelectronic materials. The workflow is fully automated, significantly reducing the number of physics-based simulations to predict materials properties in an extensive library. The rapid screening of datasets allows a better understanding of structure-function relationships for systematic design and application of optoelectronic materials with higher efficiency.””
Case Study
Recent studies by Schrödinger, published in Frontiers in Chemistry and presented at SID-Display Week 2022, have demonstrated the active learning paradigm for OLED materials discovery.1,2
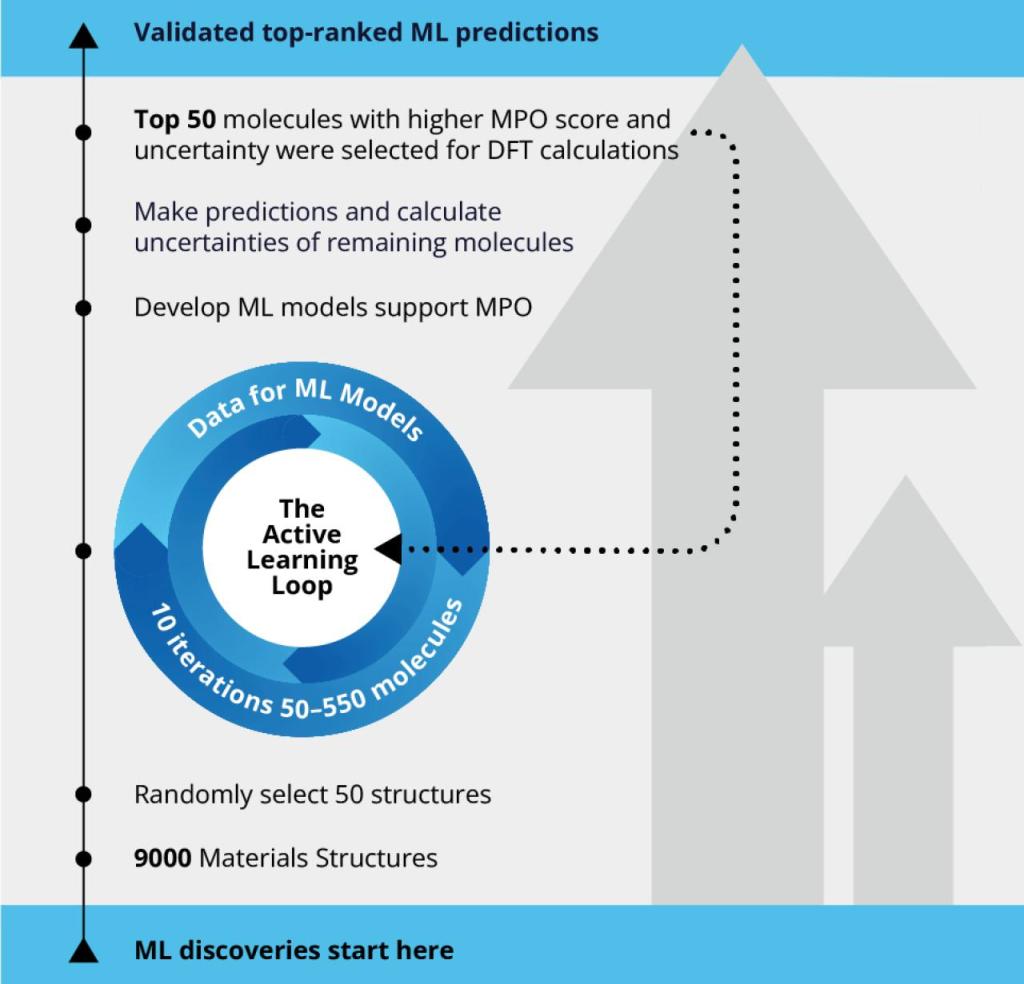
To explore the best-performing hole-transporting molecules for OLED, scientists screened a large pool of 9,000 molecules using the automated active learning workflow. The initial training set included 50 molecules for which a machine learning model was developed supporting efficient multi-parameter optimization (MPO). The ML model was then used to predict the rest of 8,950 molecules in the pool, each of which only costs a fraction of second. Top 50 molecules with higher MPO score and uncertainty were selected for DFT calculations and the calculated data were input for the next iteration of ML model training. The size of the training set for active learning was increased from 50 to 550 molecules in 10 iterations.
Active learning is the iteration of these steps until the DFT calculated data and the machine learning predicted data converge with sufficient accuracy. By using the AL approach, the scientific team was able to screen the chemical space of the materials library 18 times faster than the traditional approach of expensive quantum mechanical calculations, leading to considerable time and resource savings.
The Active Learning Workflow
References
-
Active Learning Accelerates Design and Optimization of Hole Transporting Materials for Organic Electronics Hadi Abroshan, H. Shaun Kwak, Yuling An, Christopher Brown, Anand Chandrasekaran, Paul Winget and Mathew D. Halls, Front. Chem., 2022, 9, 800371
-
Active Learning for the Design of Novel OLED Materials Hadi Abroshan, Anand Chandrasekaran, Paul Winget, Yuling An, Shaun Kwak, Christopher T. Brown, Tsuguo Morisato, and Mathew D. Halls, SID Symposium Digest of Technical Papers, 2022, 53, 885-888
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Learning to Taste: Application of Deep Learning to Predict the Sweetness of Small Organic Molecules


AUG 6, 2020
Learning to taste: Application of deep learning to predict the sweetness of small organic molecules
Speaker:
Atif Afzal, Senior Scientist
Abstract:
With the recent developments in machine learning techniques, we use the data available on the taste of existing chemistries to develop efficient data-driven models for taste prediction. We demonstrate that these advanced machine learning models are highly efficient in classifying the molecules as bitter/sweet. Using the data and the model developed in this work, we can not only predict the sweetness of new molecules but also identify underlying relationships that distinguish the molecules as bitter/sweet.
CECAM Workshop 2026

Conference
CECAM Workshop 2026
- October 14th-16th, 2026
- Lyon, France
Schrödinger is thrilled to serve on the organizing committee for the upcoming workshop, “Coarse-Graining in the Age of Machine Learning and Molecular Design” on October 14th – 16th in Lyon, France.
At the event, our team will deliver a talk, “Bridging Scales in Formulation Science: Coarse-Grained Simulations with the Schrödinger Software Suite,” showcasing how tailored CG methodologies (like DPD and Martini) accelerate product design.
Don’t miss the chance to share your work at the intersection of CG modeling and machine learning. Register or submit your abstract! Discuss your work challenges with Schrödinger scientists at the workshop.
Bridging Scales in Formulation Science: Coarse-Grained Simulations with the Schrödinger Software Suite
Speaker:
Irene Bechis, Principal Scientist I, Materials Science Applications Science
Abstract:
Coarse-grained (CG) modeling has emerged as a vital approach for probing the behavior of complex molecular systems at length- and timescales beyond the reach of conventional atomistic molecular dynamics (MD). Driven by the necessity to bridge the gap between microscopic interactions and macroscopic phenomena, the field has recently experienced rapid acceleration in both foundational method development and diverse industrial applications. To keep pace with these modern advancements, the Schrödinger software suite has continuously evolved, integrating cutting-edge CG workflows and rigorously validating them against increasingly complex and challenging systems.
This presentation showcases the value of the CG approach through case studies spanning applications in the areas of pharmaceutical formulations and consumer packaged goods. Key examples will include modeling the self-assembly, mRNA encapsulation, and delivery mechanisms of lipid nanoparticles (LNPs) and simulating how complex commercial formulations interact with, penetrate, or modify biological substrates such as hair and skin. Utilizing a range of distinct CG methodologies—specifically Dissipative Particle Dynamics (DPD) and the Martini force field framework—we demonstrate how tailored simulation workflows can accelerate product design.
Collectively, these case studies illustrate how modern CG modeling within the Schrödinger ecosystem provides critical insights that enable the rational design and optimization of next-generation therapeutic and consumer products.
Physics-driven ML to accelerate the design of layered multicomponent electronic devices

FEB 10, 2026
Physics-driven ML to accelerate the design of layered multicomponent electronic devices
Many advanced electronic devices – such as OLEDs, batteries, solar cells, and transistors – rely on complex multilayer architectures composed of multiple materials. Optimizing device performance, stability, and efficiency requires precise control over layer composition and arrangement, yet experimental exploration of new designs is costly and time-intensive. Although physics-based simulations offer insight into individual materials, they are often impractical for full device architectures due to computational expense and methodological limitations.
Schrödinger has developed a machine learning (ML) framework that enables users to predict key performance metrics of multilayered electronic devices from simple, intuitive descriptions of their architecture and operating conditions. This approach integrates automated ML workflows with physics-based simulations in the Schrödinger Materials Science suite, leveraging physics-based simulation outputs to improve model accuracy and predictive power. This advancement provides a scalable solution for rapidly exploring novel device design spaces – enabling targeted evaluations such as modifying layer composition, adding or removing layers, and adjusting layer dimensions or morphology. Users can efficiently predict device performance and uncover interpretable relationships between functionality, layer architecture, and materials chemistry. While this webinar focuses on single-unit and tandem OLEDs, the approach is readily adaptable to a wide range of electronic devices.
Key highlights:
- A machine learning framework for modeling electronic device performance, allowing users to define architectural features to explore novel device configurations
- Model accuracy demonstrated with a dataset of over 2,000 OLED architectures for multiple key performance metrics
- Pre-trained ML models for six device performance metrics available out-of-the box, including external quantum efficiency, current efficiency, power efficiency, electroluminescence maximum peak position, bandwidth, and emission color
- Intuitive graphical interface for designing, training, and exploring new chemistries and device architectures
- Demonstration of the framework’s extensibility to a broad range of electronic devices
Who should attend:
- Device developers
- R&D leaders
- Innovation managers
- Digitization managers
- Synthetic chemists
- Computational materials scientists
Our Speaker

Kevin Moore
Senior Scientist II, Materials Science Software, Schrödinger
Kevin Moore is a scientist at Schrӧdinger working on development of multiscale and hybrid physics-AI predictive frameworks for discovery and optimization of next generation materials, devices and fabrication. Prior to joining Schrӧdinger, he earned a Ph.D. in computational chemistry from the University of Georgia and conducted postdoctoral research at Argonne National Laboratory. He specializes in quantum physics-based calculations to predict the structure, properties, and reactivity of chemical systems. Recently, his efforts have been on training and validating new ML architectures and models, leveraging first-principles data, informatics and physics featurization. These new AI frameworks bridge chemistries and length scales, spanning from atoms to devices. One particular target area involves the design of electronic devices such as OLEDs, batteries, solar cells, transistors, and more.
Optimizing Viscosity and Cost in Formulations with Missing Structural Data
Virtual Screening Online Course FAQ

FAQ
Virtual Screening Online Course
I have registered with a start date in the future
How do I change my course start date?
You can modify your course registration here, and choose a new time frame that works for you. This is also a great page to double-check your end-dates across courses.
I am currently taking the course
How do I access the virtual cluster (i.e. how do I access Maestro)?
Your credentials for the virtual cluster that you will use to access web-based Maestro were shared in an email with the subject line “Virtual screening with integrated physics and machine learning — Welcome and Credentials!”. If you have not received that email please make sure to first check your Spam and Junk folders and let us know so we can find other ways to deliver the information. Your username with the Masetro virtual workstation will be a user number, for instance “user5”.
To access the virtual cluster, choose the geographic region that is closest to you:
North America/South America access
Europe/Africa/Asia/Australia access
Please note that each Schrödinger Online Course has a different set of virtual cluster URLs. If you have enrolled in multiple courses please be sure to use the correct course-specific URL to log into the virtual workstation for the course you are currently working on. The virtual clusters require high-speed internet for ideal performance.
Please also remember that uploading or downloading files or data to web-based Maestro that are not related to the course is strictly prohibited.
Can I have an extension?
The timing of this course has been found to work well with the schedule of a participant with a full-time role. However, we understand that schedules are dynamic and we would like to ensure all participants feel they have adequate time to complete the course modules and final assignment. Due to the nature of the computational resources that are provisioned for the course, extensions are evaluated on a case-by-case basis. Please fill out this here to request an extension.
I have completed the course
How do I access my course certificate and badge?
Your certificate and badge will be available within the course platform when you reach 100% completion for your course. Be sure to complete any unfinished lessons and be aware that assignments need to be approved by a Schrödinger scientist before an assignment lesson is considered complete.
After the course, can I continue to use Maestro for my own research?
Unfortunately, the virtual cluster is only used for our online certification courses. If you are interested in using Maestro for your own research, please reach out and we can provide you more information on how to bring Schrödinger tools to your research group.
Accelerating Product Development: The Industrial Shift to AI/ML-Driven Formulation


Accelerating Product Development: The Industrial Shift to AI/ML-Driven Formulation
Schrödinger participated in a podcast hosted by Innovation Research Interchange on September 18th.
In this discussion, we explored the rapidly evolving role of modeling and machine learning in formulation design; from a supplementary tool to a driving force of innovation. Once considered a “nice to have,” computational modeling is now helping to replace costly and time-consuming physical experimentation, and accelerating product development across industries from CPG to aerospace to semiconductor.
We discussed how advances in AI/ML and physics-based simulations are enabling researchers to tackle the growing complexity of real-world formulations, bridging the gap between theory and commercial products. Whether you’re a formulation scientist, data enthusiast, or R&D leader, this discussion sheds light on how digital tools are transforming the lab and the future.
Our Speaker

Jeffrey Sanders
Product Manager and Scientific Lead, Consumer Goods, Schrödinger
Jeff Sanders received his B.S. in applied physics from Worcester Polytechnic Institute and then his Ph.D. in biophysics and molecular pharmacology from Thomas Jefferson Medical College. Since joining Schrödinger in 2013, he has served several roles. Jeff is currently the product manager and technical lead for the consumer packaged goods applications group. Additionally, he is a managing board member of the Food Engineering, Expansion, and Development (FEED) Institute, and also holds a faculty position in the Food Science Department at UMass Amherst.
Hit discovery course bundle

Hit discovery course bundle
Includes access to all paid, intermediate life science courses, including: designing quality ligand libraries, target enablement, validation, and preparation, and virtual screening with integrated physics and machine learning
Details
Available Languages
Chinese, English, Japanese, Korean
Duration
3 months from selected start date
Level
Intermediate
Cost
$1510 for non-student users
$525 for student / post-doc
Who should take this course?
Medicinal chemists, cheminformaticians, ML scientists, new computational chemists
Overview
As structural data and ligand libraries continue to grow, so does the demand for validated, scalable, and computationally efficient hit discovery workflows.
This course bundle combines all three of Schrödinger’s intermediate life science courses into one powerful program, designed to build practical expertise across the entire virtual screening pipeline.
Through this series you’ll gain hands-on experience with Schrödinger’s industry-leading Maestro and command-line interface. Ideal for scientists looking to enhance their practical skills in structure-based modeling, ligand library design, and virtual screening techniques.
An opportunity to professionally develop, deepen drug discovery skills, and earn certifications and digital badges that demonstrate capabilities across all major stages of the target validation and hit discovery process.
- Prepare and refine protein structures, including exercises with AlphaFold structures and cryptic pocket identification
- Understand the vastness of chemical space, and design and filter ligand libraries using profiling and enumeration strategies
- Execute virtual screening campaigns with Active Learning Glide and other advanced computational tools
- Work through real-world case studies challenging your skills in each focus area
- Learn on your own schedule with expert guidance and curated learning content
This course comes with temporary access to a web-based version of Schrödinger software, complete with licenses and compute resources
Requirements
- A computer with reliable high speed internet access (8 Mbps or better)
- A mouse and/or external monitor (recommended but not required)
- Working knowledge of general chemistry
- Working knowledge of Maestro. Please work through our Getting Going with Maestro resources to become familiar with using Maestro.
- (Optional) Prior completion of the Introduction to molecular modeling in drug discovery online certification course.
Certification
- A certificate signed by the Schrödinger course lead to add to your CV or resume
- A badge that can be posted to social media, such as LinkedIn

What you will learn
Target enablement and assessment
Prepare, assess, and refine experimental (X-ray, cryo-EM) and ML-predicted (AlphaFold, homology) structures. Identify druggable binding sites and characterize cryptic pockets
Library design
Explore chemical space, profile vendor libraries, and generate tailored in silico enumerated libraries. Learn strategies to filter libraries to remove liabilities while preserving diversity
Executing and validating virtual screens
Run pilot virtual screens, prepare receptor grids, apply known active enrichment validation techniques
Hit evaluation and prioritization
Evaluate and prioritize hits, including inspection and clustering. Scale screening with machine learning and AB-FEP+
Course syllabus
The course bundle includes access to the following three courses in their entirety during the single course session.
Designing quality ligand libraries
Target enablement, preparation, & validation
Virtual screening with integrated physics & machine learning
Need help obtaining funding for a Schrödinger Online Course?
We proudly support the next generation of scientists and are committed to providing opportunities to those with limited resources. Learn about your funding options for our online certification courses as a student, post-doc, or industry scientist and enroll today!
Show off your newly acquired skills with a course badge and certificate
When you complete a course with us in molecular modeling and are ready to share what you learned with your colleagues and employers, you can share your certificate and badge on your LinkedIn profile.
Frequently asked questions
How much does the Hit discovery course bundle online course cost?
Pricing varies by each course and by the participant type. For students wishing to take this, we offer a student price of $500, and $1435 for non-students.
What time are the lectures?
Once the course session begins, all lectures are asynchronous and you can view the self-paced videos, tutorials, and assignments at your convenience. When registering for the course you will select the start and end date. Within those dates, you will have asynchronous access to the course material and virtual workstation to work on the course when it best suits your schedule.
How could I pay for this course?
Interested participants can pay for the course by completing their registration and using the credit card portal for an instant sign up. Please note that a credit card is required as we do not accept debit cards. Additionally, we can provide a purchase order upon request, please email online-learning@schrodinger.com if you are interested in this option. If you have any questions regarding how to pay for the course, please visit our funding options page.
How can I preview the course before registering?
Are there any scholarship opportunities available for students?
Schrödinger is committed to supporting students with limited resources. Schrödinger’s mission is to improve human health and quality of life by transforming the way therapeutics and materials are discovered. Schrödinger proudly supports the next generation of scientists. We have created a scholarship program that is open to full-time students or post-docs to students who can demonstrate financial need, and have a statement of support from the academic advisor. Please complete the application form if you qualify for our scholarship program!
Will material still be available after a course ends?
While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course.
Do I need access to the software to be able to do the course? Do I have to purchase the software separately?
For the duration of the course, you will have access to a web-based version of Maestro, Bioluminate, Materials Science Maestro and/or LiveDesign (depending on the course). You do not have to separately purchase access to any software. While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course. Please note that Schrödinger software is only to be used for course-related purposes.
Related courses
 Life Science
Life Science
Life Science
Life Science
Target enablement, preparation, & validation
Enabling protein structures from x-ray crystallography, cryo-EM, ML-methods, and homology modeling for structure-based computational workflows
 Life Science
Life Science
Life Science
Life Science
Designing quality ligand libraries
Exploring chemical space, profiling and tailoring ligand libraries, validating docking models, and methods of enumeration for hit discovery
 Life Science
Life Science
Life Science
Life Science
Virtual screening with integrated physics & machine learning
Acquire essential skills in next-generation virtual screening, integrating physics and machine learning for smarter hit identification
What our alumni say