Formulation ML
Computational drug design and chemo-informatics: a hands-on course at the University of Antwerp


JUN 15, 2023
Computational drug design and chemo-informatics: a hands-on course at the University of Antwerp
The University of Antwerp is the third-largest university in the Dutch-speaking region of Belgium, with over 20,000 students annually. Within the Biochemistry and Biotechnology curriculum, students have the option to take a three-ECTS course on computational drug design and chemo-informatics. The course is organized in a modular fashion and covers both theoretical and practical sessions.
During the theoretical sessions, students learn about chemo-informatics and virtual screening, which includes concepts such as chemical fingerprints, molecular similarity, clustering, machine learning models, and virtual screening performance metrics. The course also covers molecular docking and pharmacophore searching. The concepts covered in the theoretical sessions are then put into practice in a series of hands-on sessions.
For the chemo-informatics tasks, the students use Google Colab with RDKit as a chemo-informatics toolkit, while for the pharmacophore and docking-related aspects, they use Maestro, Phase, and Glide. These tools are made available through the “”Teaching with Schrödinger”” web-based virtual workstations, which allows students to access them from anywhere at any time. Finally, using an internally-developed virtual reality system, the students can graphically study the non-bonded interactions between ligand and protein.
At the start of the course, a drug design project is defined based on ongoing research programs in the Faculty. The goal of the project is to identify a limited number of commercially-available compounds (5-10) that are subsequently purchased and biochemically characterized for their inhibitory properties. The students complete the program with a written report, which serves as the basis for the oral examination at the end.
Our Speaker

Hans De Winter
University of Antwerp
Hans De Winter was appointed in 2013 as a professor of Computational Drug Design at the University of Antwerp (Belgium) after a long career in industry, first as a senior scientist at Johnson & Johnson in Beerse, Belgium, and subsequently as a co-founder and CSO of Silicos NV. He holds a PhD from the University of Leuven (Belgium) and completed post-doctoral stays at the Victorian College of Pharmacy (Australia) and the Rega Institute in Leuven (Belgium) before starting his career as a scientist in the pharmaceutical industry. Despite his elaborated industrial background during a period of more than 20 years, he has over 60 scientific publications and is listed as inventor on eight granted patents. Hans’ research interests are mainly situated in the field of computational medicinal chemistry and cheminformatics. Current research activities include: 1) molecular dynamics-based modeling of protein-protein interactions and unraveling the kinetics of lipid membrane-bound protein complexes using large-scale molecular dynamics calculations with Markov chain modeling; 2) the elaboration of spectrophore-based algorithms for exploration of ligand/protein interaction space; 3) the development of automated in silico ligand design systems. Additional focus points are the development of open-source and cloud-based interrogation software for large chemical and biological data repositories. Finally, his expertise in in silico drug design is used in several ongoing targeted drug discovery projects. Prof. Hans De Winter is the coordinating promoter or co-promoter of a number of PhDs and has a broad research network both in industry and in academia based on his expertise in modeling/chemoinformatics. He is also chairman of the Flemish computational chemistry division of the European Association of Chemical and Molecular Sciences (EuCheMS) since 2015. He teaches ‘organic chemistry’ to 1st year bachelor students in Pharmaceutical Sciences, and ‘computational drug design and cheminformatics’ to 1st year master students in Biochemistry at the University of Antwerp.
Maestro
DeepAutoQSAR
DeepAutoQSAR
MS Formulation ML

MS Formulation ML
Automated machine learning solution to generate accurate formulation-property relationships and screen new formulations with desired properties

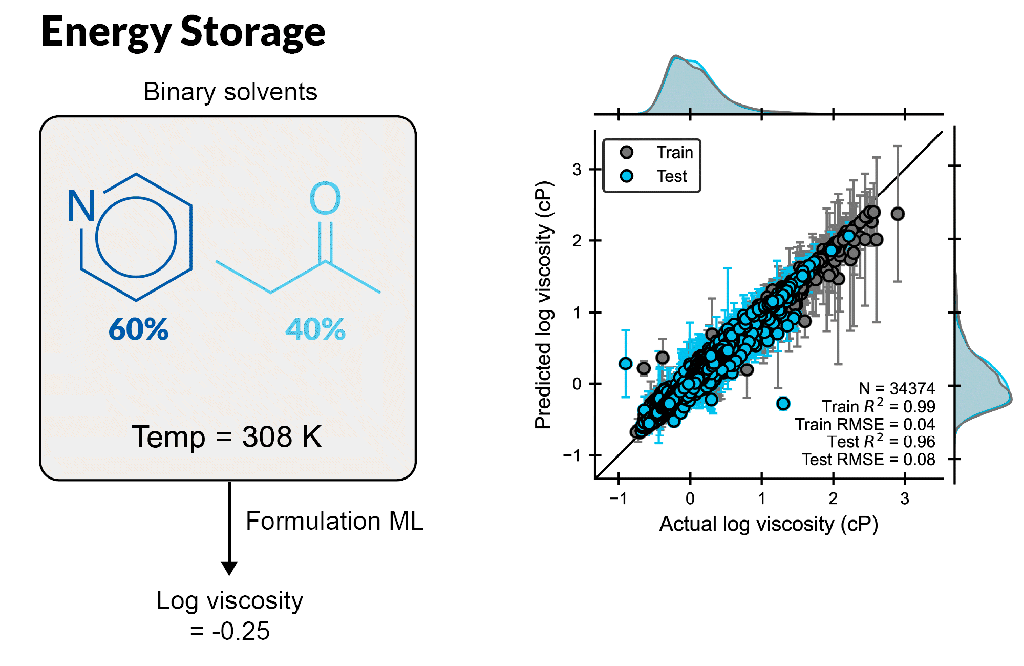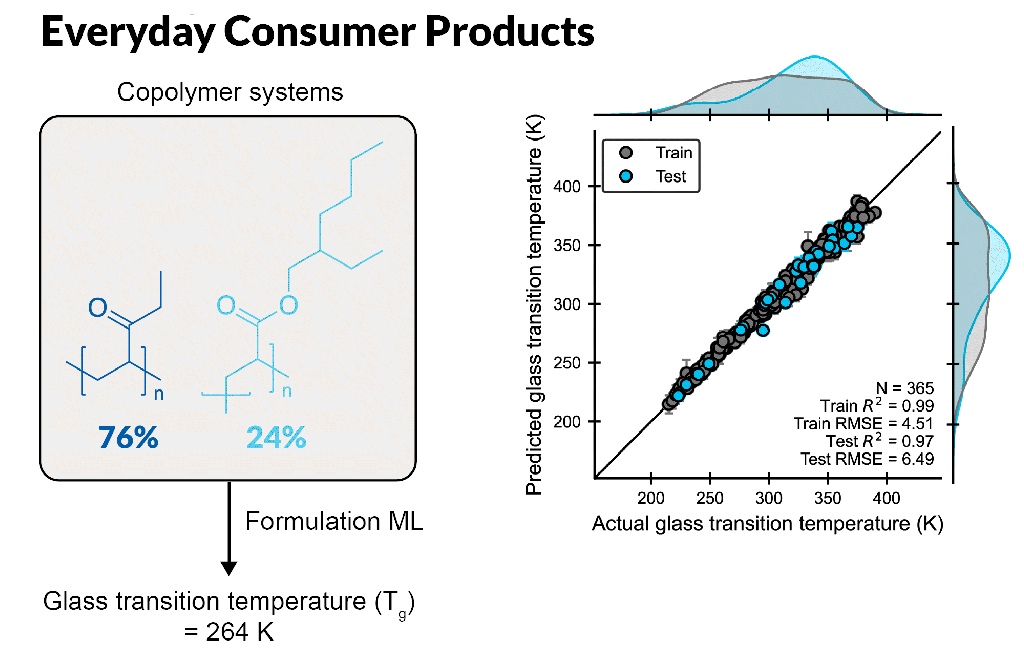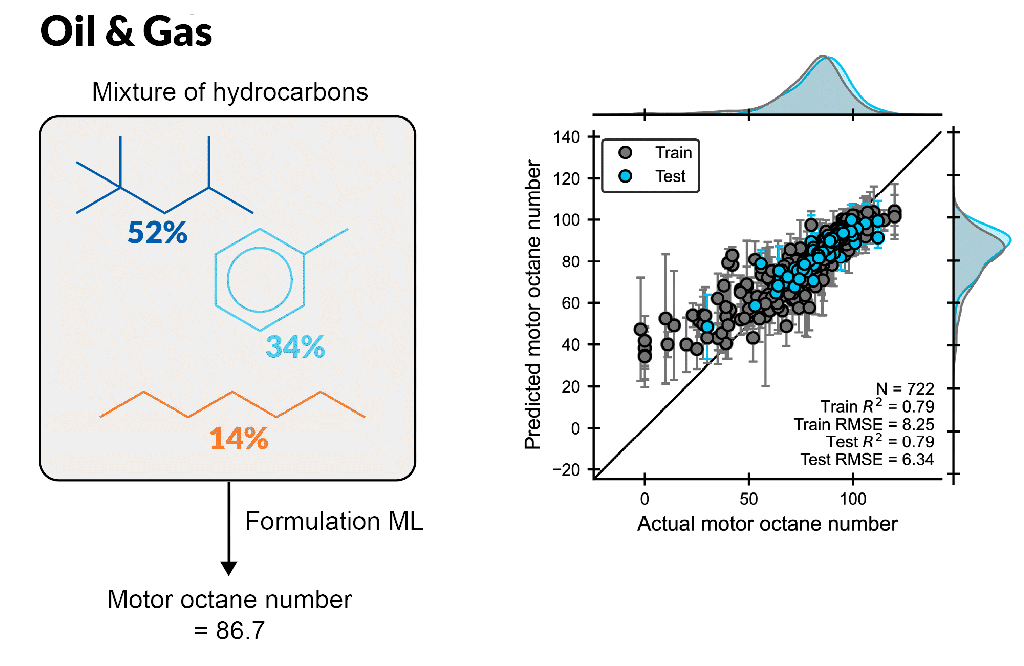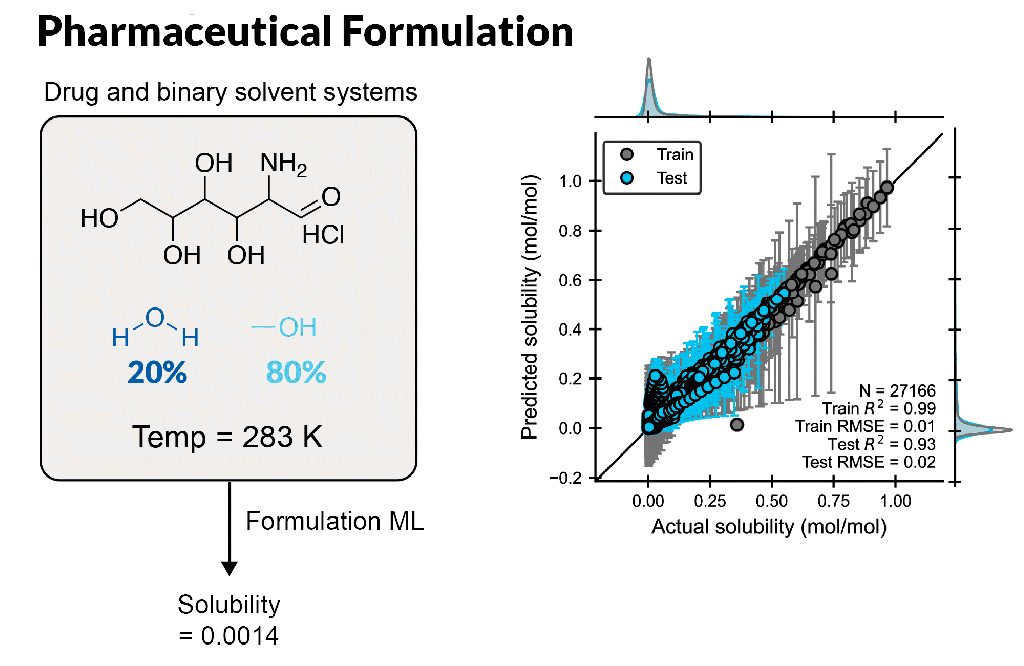
Create accurate machine learning models to design better formulations
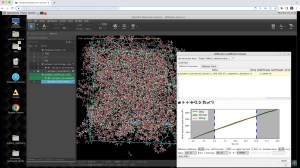
Formulation ML allows scientists to predict properties based on ingredient structures and compositions. Whether you are a formulation expert or just learning in this area, this automated, supervised learning solution enables you to gain deeper insight into formulation-property relationships.
Key Capabilities

Build formulation-property models for chemical mixtures with varying ingredient structures and compositions, which are scalable up to 100 ingredients or more
Rapidly predict novel formulations with new chemistry and composition, requiring only seconds per formulation
Understand which molecular features to focus on to fine-tune properties, leveraging feature importance tools to identify key descriptors for a property using a trained model
Enable accurate ML model development using expert cheminformatic descriptors and automatic hyperparameter tuning with minimal ML expertise
Input customized descriptors, including experimental data, in CSV format into the ML model to improve model performance
Optimize multiple properties simultaneously by modulating ingredient structure and compositions with trained ML models, providing suggestions of best formulations for the next experiment
Featured Resources
 White Paper
Materials Science
Webinar
Materials Science
White Paper
Materials Science
Webinar
Materials Science
AI/ML meets physics-based simulations: A new era in complex materials design
In this webinar, we demonstrate the application of this combined approach in designing materials and formulations across diverse materials science applications, from battery electrolytes and fuel mixtures to thermoplastics and OLED devices.
 Webinar
Life Science
Materials Science
Webinar
Life Science
Materials Science
Accelerating pharmaceutical formulations using machine learning approaches
In this webinar, we will demonstrate how Schrödinger’s integrated ML- and physics-based approaches are transforming pharmaceutical formulation design.

Broad applications across materials science research areas


Related Products
Desmond
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
Jaguar
Quantum mechanics solution for rapid and accurate prediction of molecular structures and properties
DeepAutoQSAR
Automated, scalable solution for the training and application of predictive machine learning models
MS Informatics
Automated machine learning tools for materials science applications
MS Force Field Applications
Cutting-edge force field technologies for accurate property predictions
Publications
Leveraging high-throughput molecular simulations and machine learning for the design of chemical mixtures, Alex, C., et al. npj Comput Mater 11, 72, 2025, https://doi.org/10.1038/s41524-025-01552-2.
Schedule a consultation on Schrödinger’s Formulation ML
Contact us today to explore how you can leverage advanced simulation and AI/ML to transform formulation decisions and gain competitive advantage in your industry.
Don’t see your areas of interest in the current lists above? Reach out so we can help.
Form submitted
Thank you, we’ll be in touch soon.
Software & services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.
OLED Device ML
OLED Device ML
Machine learning solution to investigate relationships between the architecture and performance of OLED devices for accelerated screening
Create machine learning models to enable high-throughput design and optimization of OLED devices
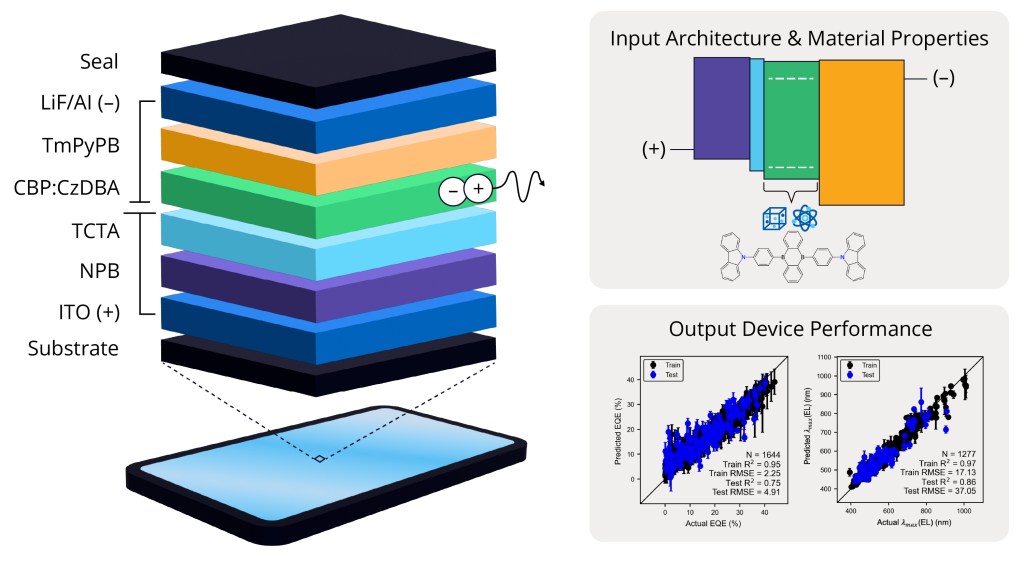
The OLED Device ML solution enables scientists to predict performance metrics that quantify the operational output, efficiency, and stability of multicomponent layered organic light-emitting diodes (OLEDs). These predictions are based upon simple and direct descriptions of device operation and architecture, such as the arrangement and chemical composition of layers. This offers a scalable solution for OLED developers seeking to perform targeted evaluations of device capability across novel design spaces.
Key Capabilities
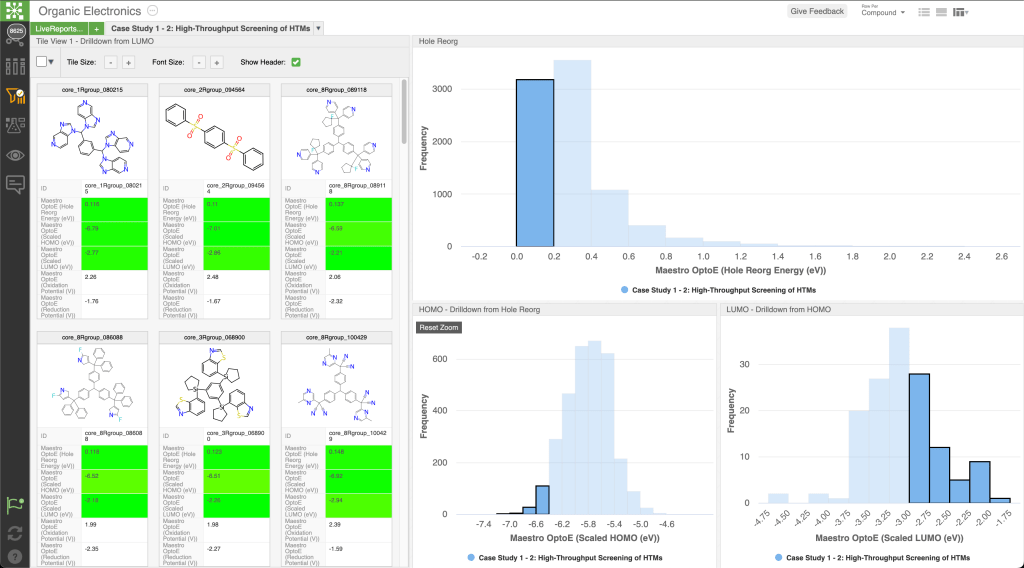
Train chemistry-informed ML models to predict performance properties for OLED devices with varying layer arrangements and chemical compositions
Rapidly predict the performance of novel device structures to establish interpretable relationships between functionality and layer architectures and chemistry
Use pre-trained ML models to predict six different device performance metrics including external quantum efficiency, current efficiency, power efficiency, electroluminescence maximum peak position, electroluminescence bandwidth, and color of the emitted light
Benefit from an intuitive graphical interface that allows easy design and exploration of novel chemistry and device architectures, facilitated by the visualization of energy level diagrams with out-of-the-box QM descriptors and ML models
Broad applications across materials science research areas
Related Products
MS Informatics
Automated machine learning tools for materials science applications
DeepAutoQSAR
Automated, scalable solution for the training and application of predictive machine learning models
Schedule a consultation on Schrödinger’s OLED Device ML
Contact us today to explore how you can leverage advanced simulation and AI/ML to design better electronic devices.
Don’t see your areas of interest in the current lists above? Reach out so we can help.
Form submitted
Thank you, we’ll be in touch soon.
Software & services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Schrödinger デジタル創薬セミナー: Into the Clinic ~計算化学がもたらす創薬プロセスの変貌~ 第14回

NOV 19, 2024
Schrödinger デジタル創薬セミナー 14:
Advancements in machine learning enhanced in silico design: Impact on a pipeline of drug discovery programs
分子特性のシミュレーションは、物理ベースのアプローチを使用することで、構造と特性の関係に関する洞察を提供し、新薬の設計を支援する分野で長らく成功を収めてきました。近年では、AIや機械学習(ML)が物理ベースのモデリング技術と組み合わさり、革新の加速に大いに貢献しています。物理ベースのモデリングの精度と一般化能力が、AI/MLモデルのパフォーマンスを向上させ、データが少ない領域でも効果的に使用できるようにしています。逆に、AI/MLのスピードと柔軟性は、物理ベースのモデルが抱える時間的・空間的な限界を克服する手助けをし、予測精度と計算効率の両方を最適化する相乗効果を生み出します。
このウェビナーでは、機械学習を活用して創薬プログラムを推進する、以下の応用例について議論します。
- FEP+を使用したアクティブラーニングによる、大規模なインシリコフラグメントスクリーニングでのヒット探索
- インテリジェントな分子コア設計のためのde novoデザインワークフローの適用
- インタラクティブなMLダッシュボードを用いたリード最適化におけるADMETプロファイルの強化のための実験データの活用
Our Speaker

Karl Leswing
Vice President Machine Learning, Schrödinger
Karl Leswing is the Vice President for Machine Learning at Schrödinger. In this role he oversees the research and execution of machine learning applications for Schrödinger’s digital chemistry platform. In 2017 he was a visiting researcher at the Pande Lab working on using deep learning techniques for drug discovery. During that time he co-authored MoleculeNet, a benchmarking paper analyzing machine learning techniques for chemoinformatics. Karl received his undergraduate degree from the University of Virginia, and a Master’s in machine learning from Georgia Tech.
Homogeneous catalysis & reactivity
Homogeneous catalysis & reactivity
Molecular quantum mechanics and machine learning approaches for studying reactivity and mechanism at the molecular level
Details
Available Languages
Chinese, English, Japanese, Korean
Duration
6 weeks / ~25 hours to complete
Level
Introductory
Cost
$575 for non-student users
$150 for student / post-doc
Course Timeframe
When registering for the course, you will be able to choose your preferred start and end date. Within those dates, you will have asynchronous access to the course to work on your preferred schedule
Overview
Computational molecular modeling tools have proven effective in materials science research and development. Chemists, physicists and engineers working in materials science will increasingly encounter molecular modeling throughout their careers, making it critical to have a foundational understanding of the cutting edge tools and methods. These courses are ideal for those who wish to develop professionally and expand their CV by earning certification and a badge.
These computational chemistry courses offer an effective and efficient approach to learn practical computational chemistry for materials science:
- Work hands-on with Schrödinger’s industry-leading Materials Science Maestro software
- Jump start your research program by learning methods that can be directly applied to ongoing projects
- Learn topics ranging from density functional theory (DFT) to molecular dynamics to machine learning for materials design
- Perform a completely independent case study to demonstrate mastery of the course content
- Benefit from review and feedback from Schrödinger Education Team experts for course assignments and course-related queries
- Work on the course materials on your own schedule whenever convenient for you
This course comes with access to a web-based version of Schrödinger software with the necessary licenses and compute resources for the course:
Requirements
- A computer with reliable high speed internet access (8 Mbps or better)
- A mouse and/or external monitor (recommended but not required)
- Working knowledge of general chemistry
Certification
- A certificate signed by the Schrödinger course lead
- A badge that can be posted to social media, such as LinkedIn

What you will learn
MS Maestro interface
Learn how to use an industry-leading interface for materials science modeling. No coding or scripting required to run modeling workflows
Density functional theory
Learn to apply DFT for automated property prediction for organic and inorganic molecules
Reaction mechanism elucidation
Learn to leverage quantum mechanical workflows to predict reaction pathways and energetics
Machine learning
Learn to apply machine learning for rapid and accurate property prediction of organic molecules and catalytically active complexes
Modules
Module 1
2 Hours
Introduction to materials modeling

Video
Introduction to materials modeling and this online course

Video tutorial
Introduction to materials science (MS) Maestro
Video
Modeling for homogeneous catalysis and reactivity

Honor code agreement and checkpoint
Module 2
7 Hours + Compute Time
Molecular quantum mechanics
Video
Introduction to molecular quantum mechanics (mQM)

Tutorials
- Functionals, basis sets and geometry optimizations
- R-group enumeration
- QM multistage workflows
- Rigid and relaxed coordinate scans
- Energies of reactions
- Organometallic complexes
End of module checkpoint
Module 3
6 Hours + Compute Time
Molecular quantum mechanics
Tutorials
- Bond and ligand dissociation energy
- Beta elimination reactions
- Locating transition states: Part 1
- Locating transition states: Part 2
- Reaction workflow for polyethylene insertion
- Nanoreactor
- Design of asymmetric catalysts with automated reaction workflow
End of module checkpoint
Module 4
3 Hours + Compute Time
Machine learning
Video
Introduction to machine learning (ML)
Tutorials
- Machine learning for materials science
- Machine learning for homogeneous catalysis
End of module checkpoint
Module 5
2 Hours + Compute Time
Guided case study
Tutorials
- Fundamental organometallic reactivity
- Combining AutoTS and reaction workflow
End of Module Checkpoint
Module 6
4 Hours + Compute Time
Independent case study

Assignment
Predicting regioselectivity of hydroboration
Course completion and certification
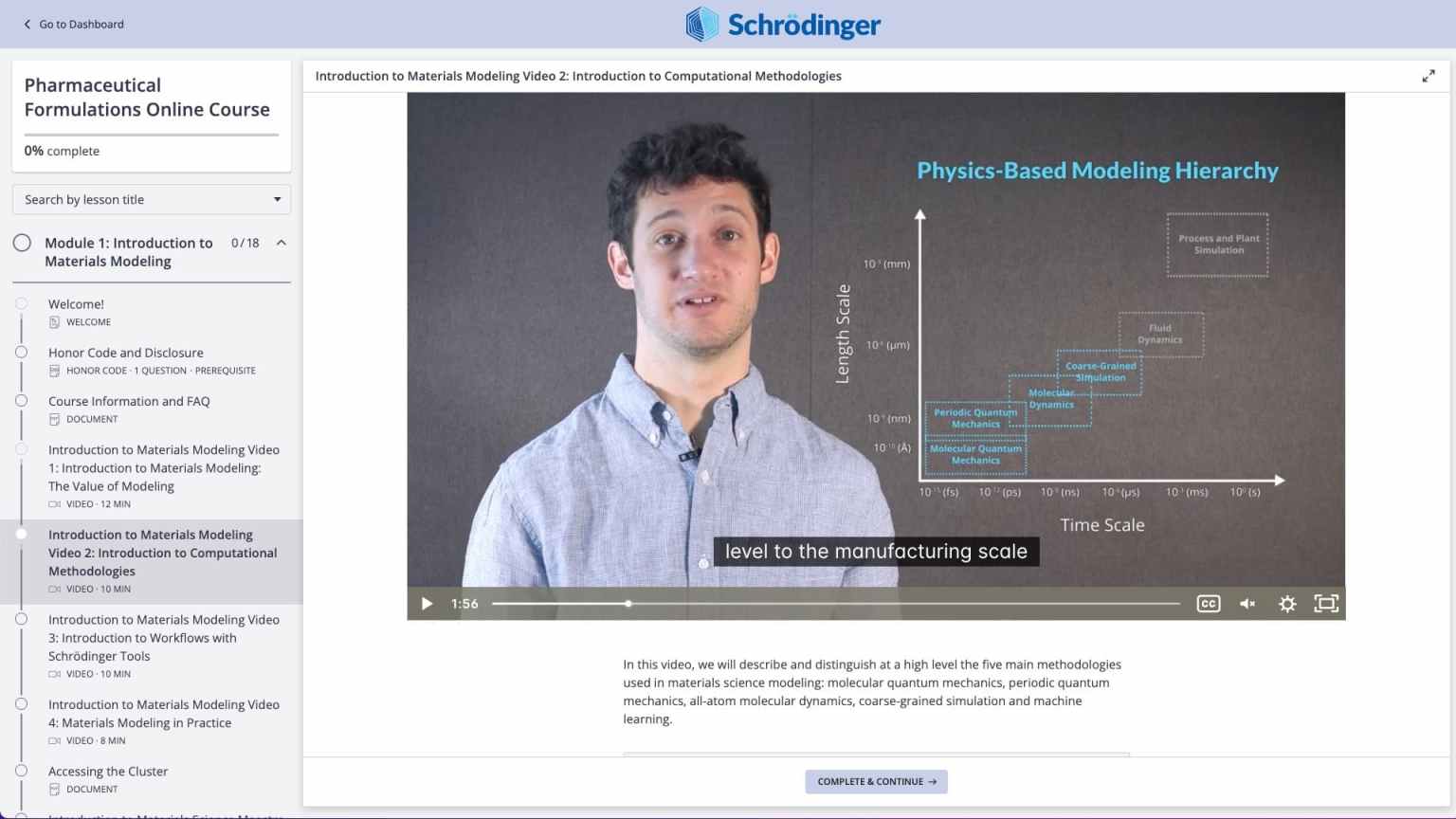
Self-paced video lessons on materials modeling
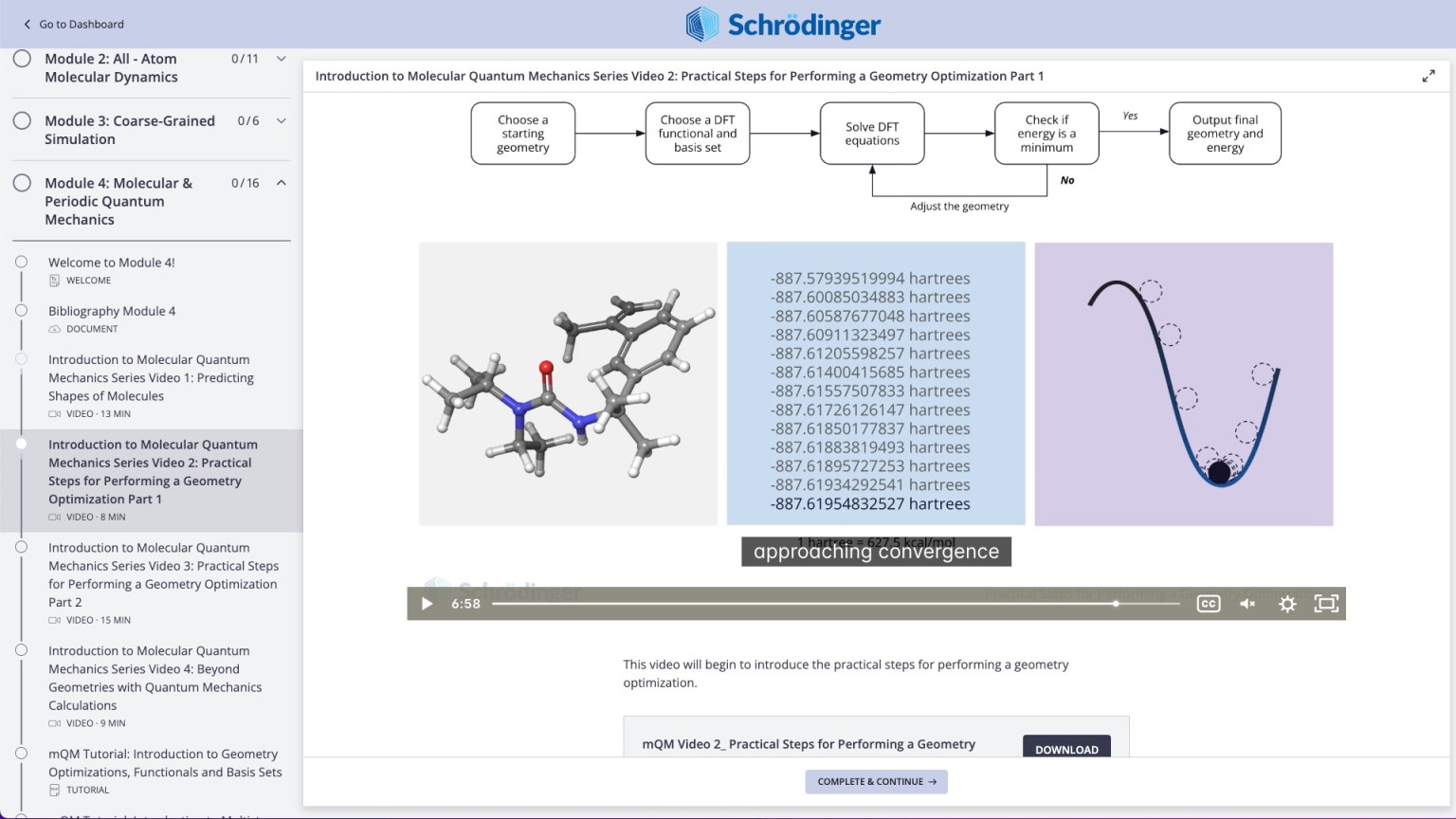
Videos on practical theory break down complex scientific concepts (e.g. Molecular Quantum Mechanics)
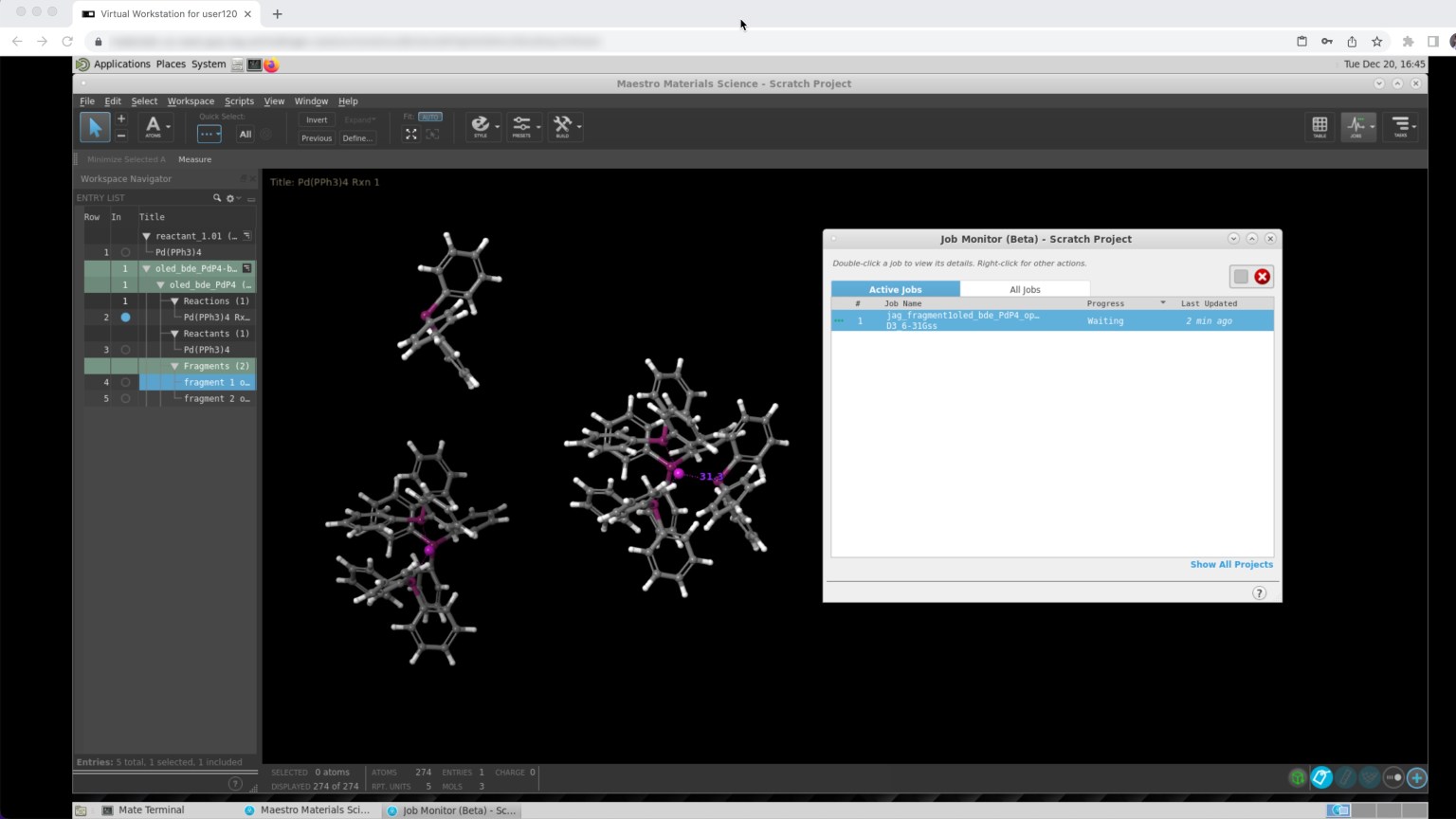
Access cloud-based computing resources to perform calculations yourself
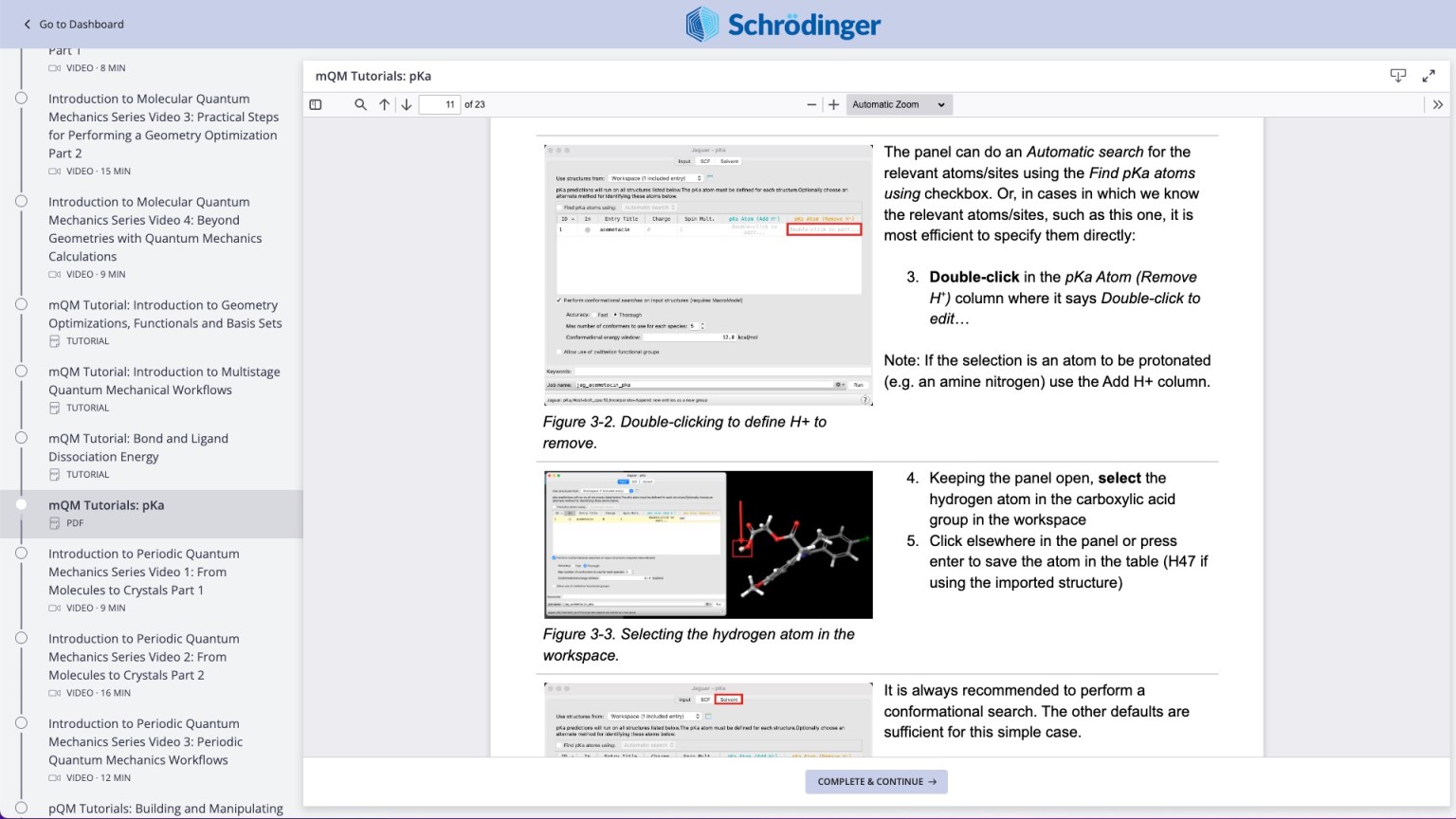
Hands-on step-by-step tutorials (e.g. Pharmaceutical Formulations course, pKa prediction)
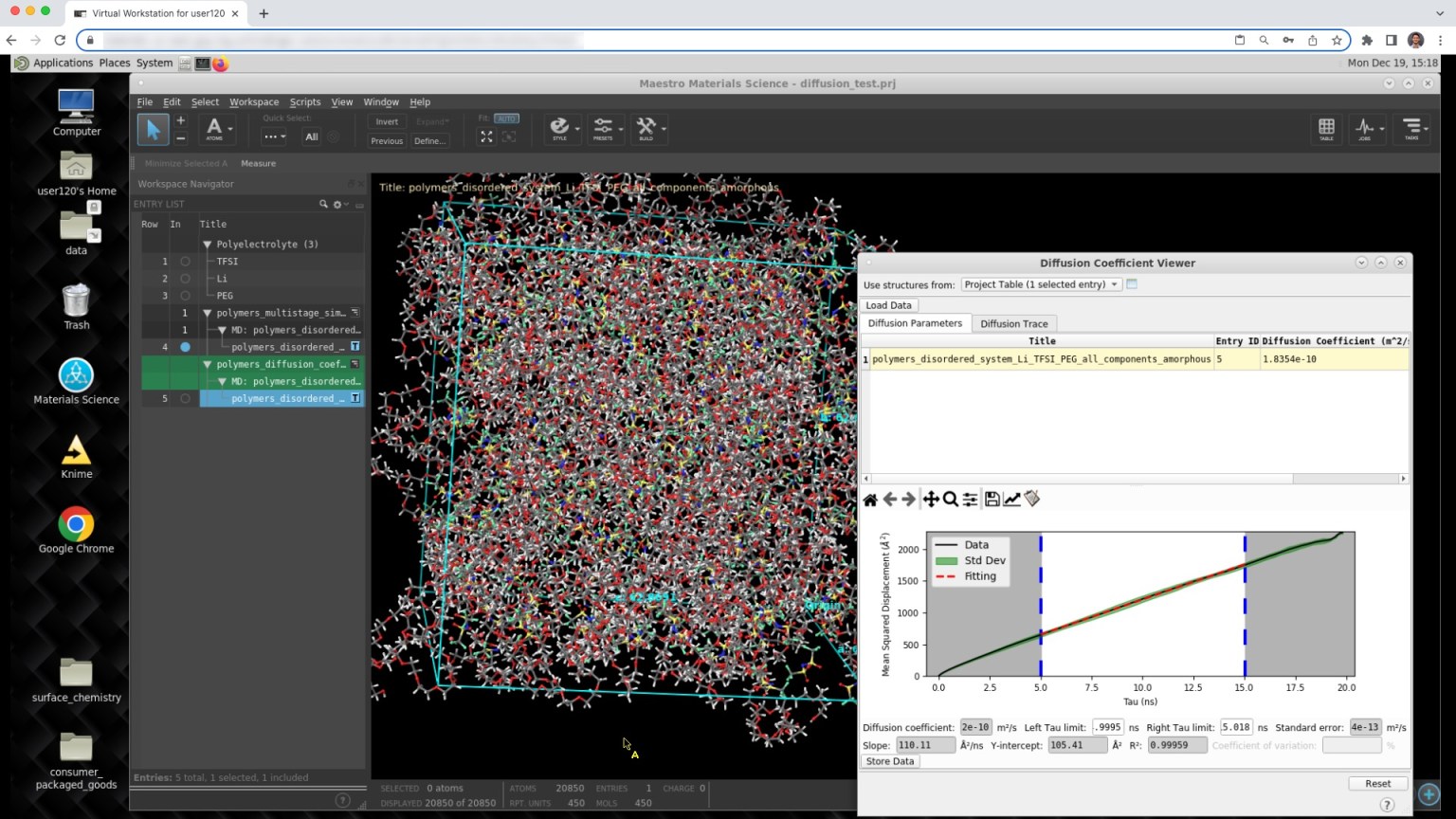
Hands-on modeling in the web-based graphical user interface (e.g. Polymeric Materials course, Diffusion tutorial)
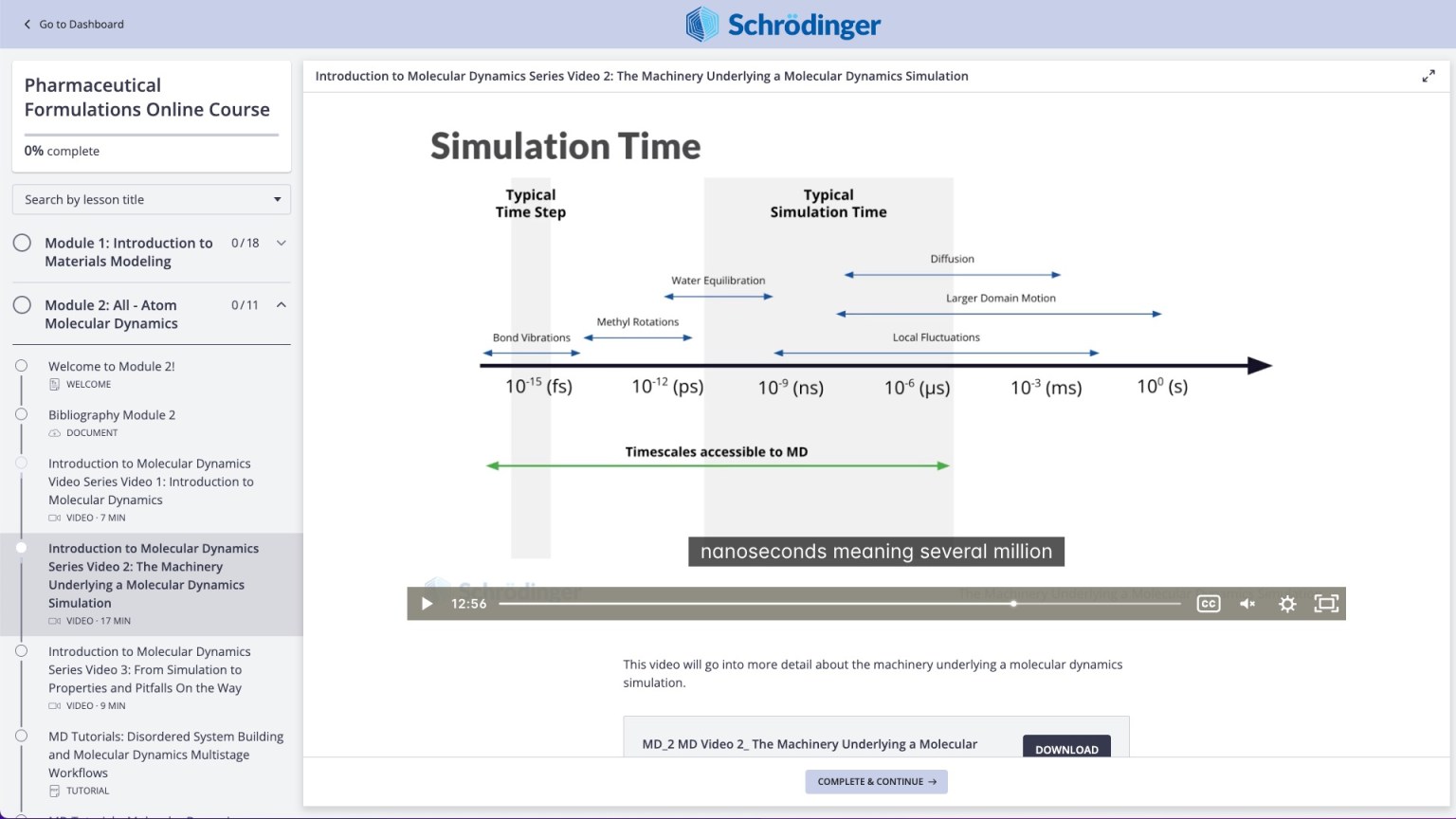
Videos on practical theory break down complex scientific concepts (e.g. Molecular Dynamics)
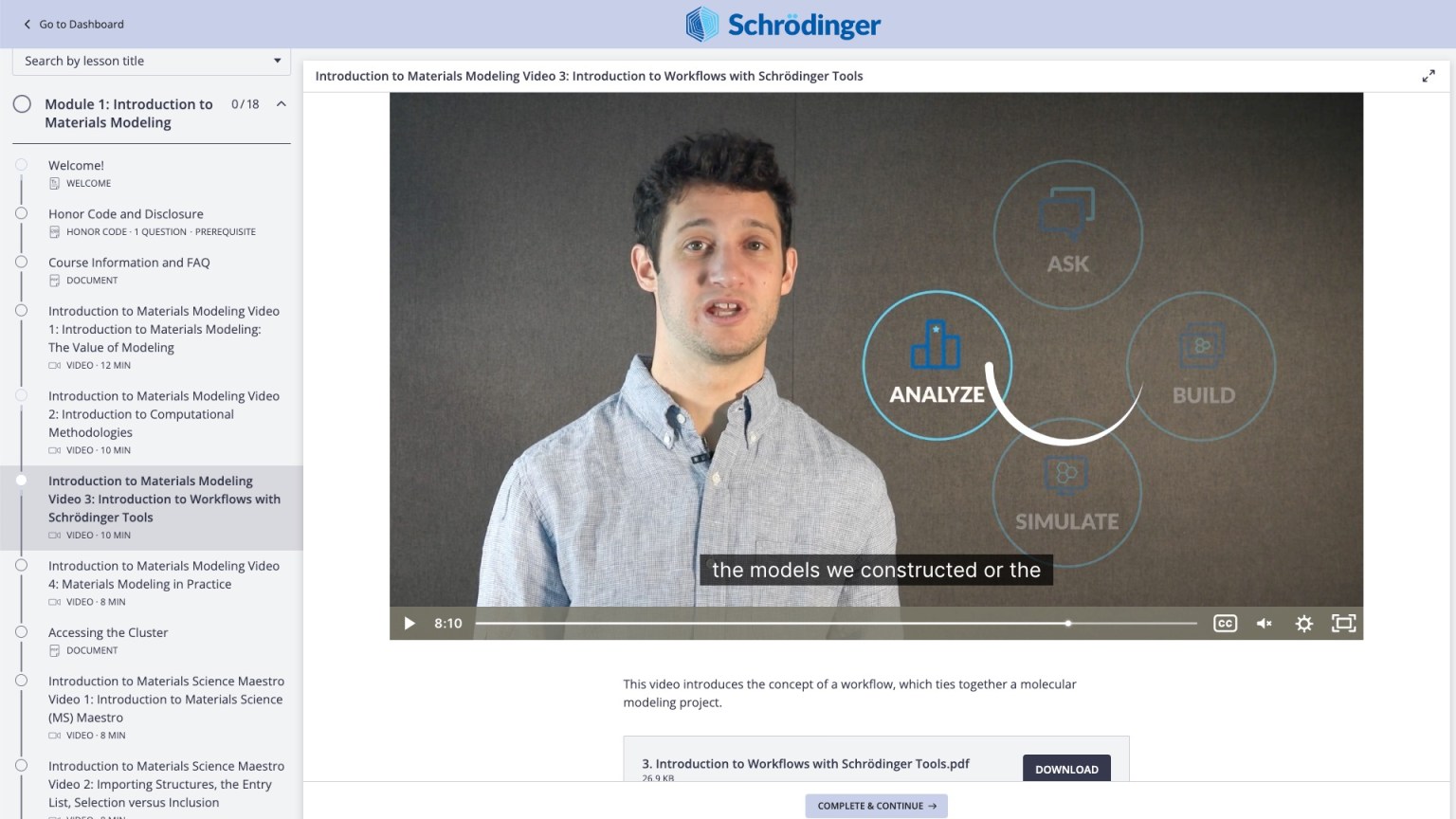
On-demand video lessons on materials modeling
Access cloud-based computing resources to perform calculations yourself
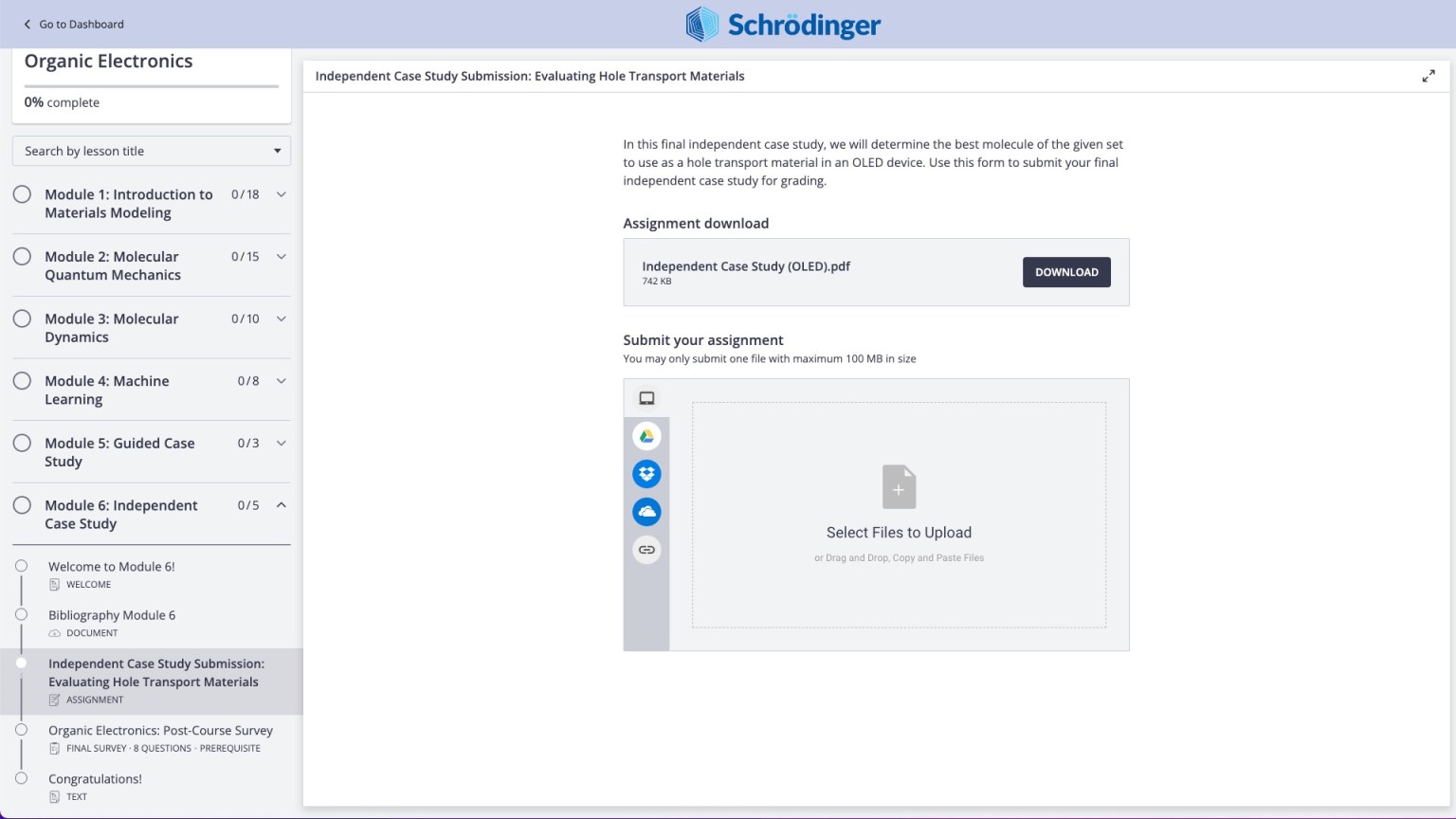
Perform case studies with expert feedback (e.g. Organic Electronic Course, Independent Case Study)
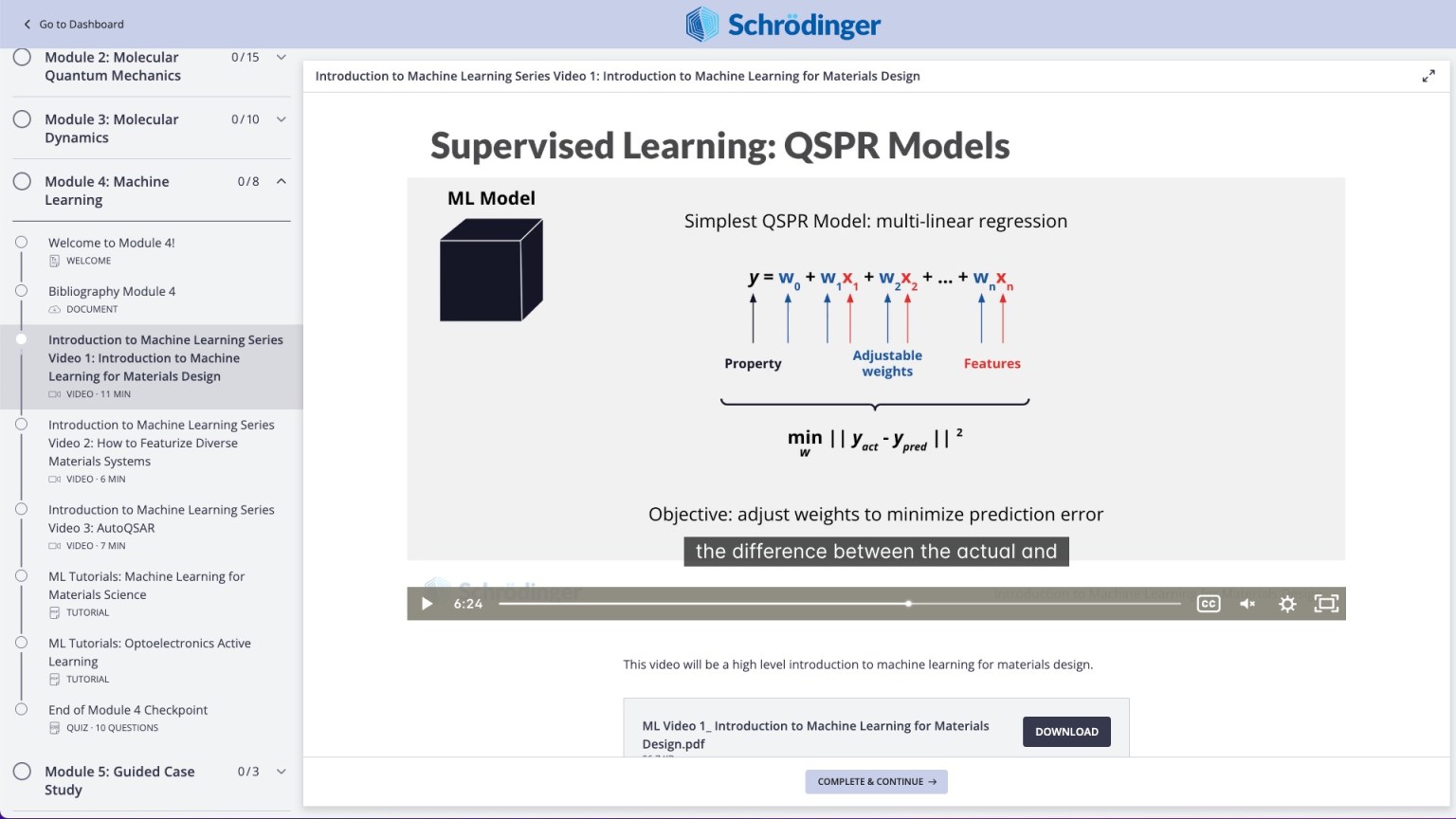
Video on practical theory break down complex scientific concepts (e.g. Machine Learning for Chemistry)
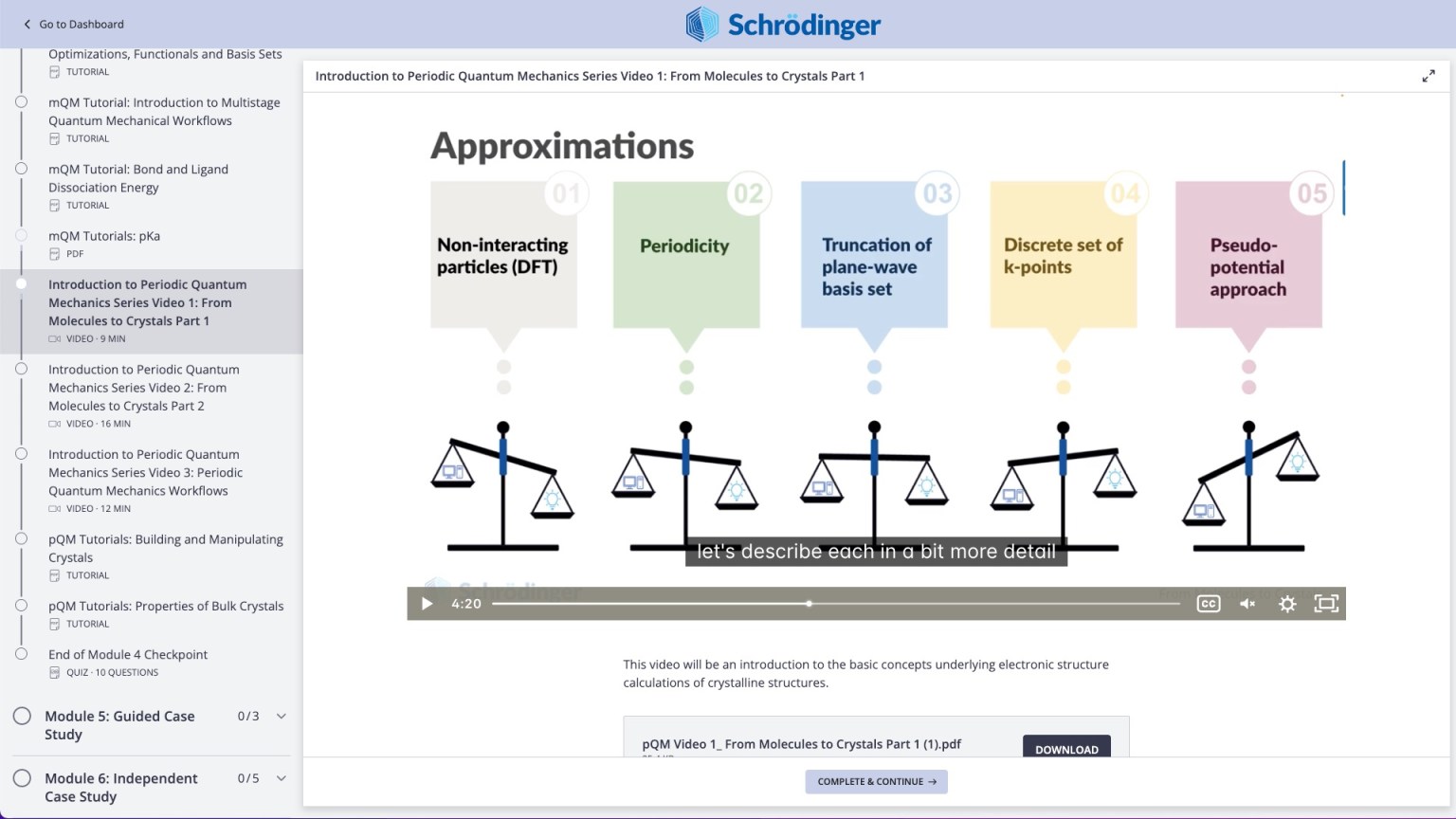
Videos on practical theory break down complex scientific concepts (e.g. Periodic Quantum Mechanics)
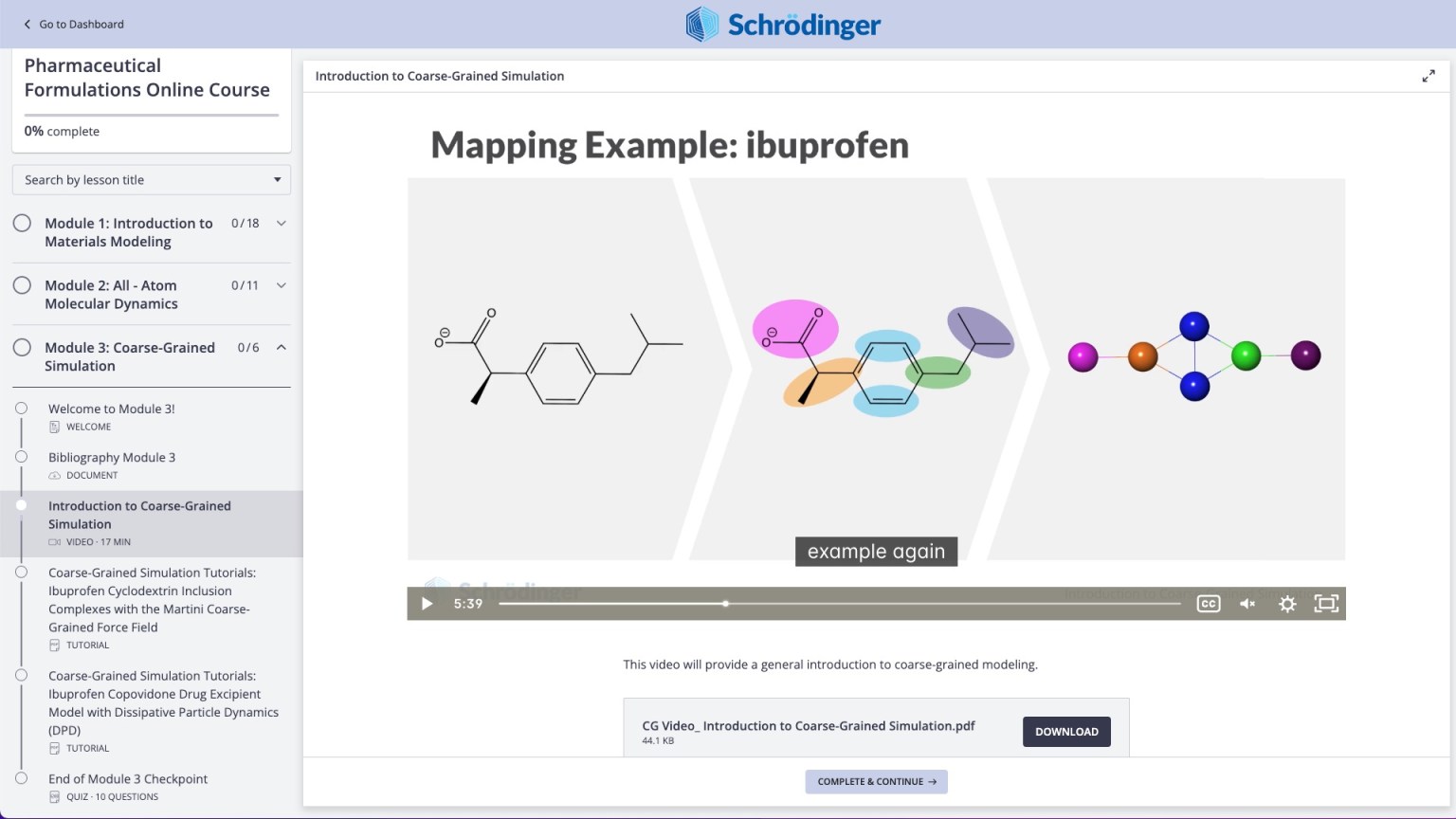
Videos on practical theory break down complex scientific concepts (e.g. Coarse-Graining)

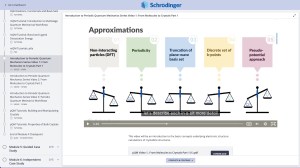
Need help obtaining funding for a Schrödinger Online Course?
We proudly support the next generation of scientists and are committed to providing opportunities to those with limited resources. Learn about your funding options for our online certification courses as a student, post-doc, or industry scientist and enroll today!
Show off your newly acquired skills with a course badge and certificate
When you complete a course with us in molecular modeling and are ready to share what you learned with your colleagues and employers, you can share your certificate and badge on your LinkedIn profile.
Frequently asked questions
How much do the online courses cost?
Pricing varies by each course and by the participant type. For students wishing to take these courses, we offer a student price of $150 for introductory courses, $305 for the Materials Science bundle, and $870 for advanced courses. For commercial participants, the course price is $575 for introductory courses and $1435 for advanced courses and bundles.
When does the course start?
The courses run on sessions, which range from 3-6 week periods during which the course and access to software are available to participants. You can find the course session and start dates on each course page.
What time are the lectures?
Once the course session begins, all lectures are asynchronous and you can view the self-paced videos, tutorials, and assignments at your convenience.
How could I pay for this course?
Interested participants can pay for the course by completing their registration and using the credit card portal for an instant sign up. Please note that a credit card is required as we do not accept debit cards. Additionally, we can provide a purchase order upon request, please email online-learning@schrodinger.com if you are interested in this option. If you have any questions regarding how to pay for the course, please visit our funding options page.
How can I preview the course before registering?
Are there any scholarship opportunities available for students?
Schrödinger is committed to supporting students with limited resources. Schrödinger’s mission is to improve human health and quality of life by transforming the way therapeutics and materials are discovered. Schrödinger proudly supports the next generation of scientists. We have created a scholarship program that is open to full-time students or post-docs to students who can demonstrate financial need, and have a statement of support from the academic advisor. Please complete the application form if you qualify for our scholarship program!
Will material still be available after a course ends?
While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course.
Do I need access to the software to be able to do the course? Do I have to purchase the software separately?
For the duration of the course, you will have access to a web-based version of Maestro, Bioluminate, Materials Science Maestro and/or LiveDesign (depending on the course). You do not have to separately purchase access to any software. While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course. Please note that Schrödinger software is only to be used for course-related purposes.
Related Courses
 Materials Science
Materials Science
Materials Science
Materials Science
Polymeric materials
All-atom molecular dynamics and machine learning approaches for studying polymeric materials and their properties under various conditions
 Materials Science
Materials Science
Materials Science
Materials Science
Course bundle
Access all materials science courses with a single, discounted registration
 Materials Science
Materials Science
Materials Science
Materials Science
Pharmaceutical formulations
Molecular and periodic quantum mechanics, all atom molecular dynamics, and coarse-grained approaches for studying active pharmaceutical ingredients and their formulations
Supporting Associations
Schrödinger デジタル創薬セミナー: Into the Clinic ~計算化学がもたらす創薬プロセスの変貌~
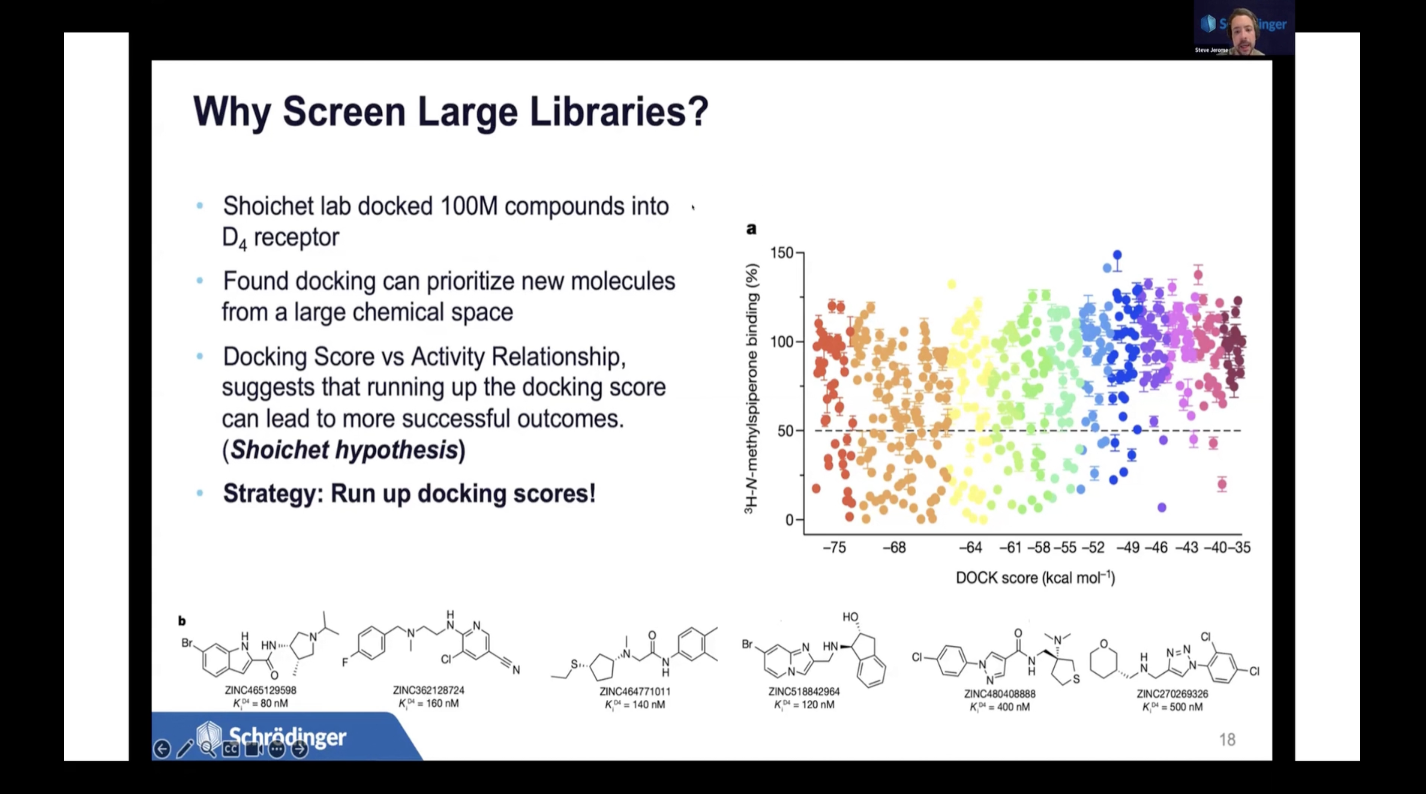
MAR 28, 2024
Schrödinger デジタル創薬セミナー:
Modern Virtual Screening Technologies that Actually Deliver High-Quality, Developable Hits
従来のバーチャルスクリーニング手法は、ヒット率の低さや新規性の欠如、そして特定した分子の開発可能性の低さに悩まされてきました。
しかし、シュレーディンガーは、AI/Machine LearningによるアクティブラーニングとAbsolute Binding FEP+を活用した新しいバーチャルスクリーニング手法を開発し、超大規模な化学ライブラリを効果的にスクリーニングすることができるようになりました。性能は大幅に向上し、複数のターゲットにおいて二桁のヒット率を達成しています。
本講演では、この高性能バーチャルスクリーニング手法を用いた、弊社の最新の創薬研究事例を紹介します。
Our Speaker

Steven Jerome
Senior Director
コロンビア大学で化学の博士号を取得。現在はヒットディスカバリー部門のディレクターとして、小分子ヒット同定のための計算ツール開発を指揮しています。
Beyond AI: The importance of physics-based simulations in next generation food design


MAY 9, 2024
Beyond AI: The importance of physics-based simulations in next generation food design
Schrödinger will be presenting in a live webinar on Beyond AI: The importance of physics-based simulations in next generation food design. This virtual event will be hosted by IFT (Institute of Food Technologists) on May 9th and features Dr. Jeffrey Sanders, product manager at Schrödinger.
Attend this webinar and learn:
- How to leverage data from physics-based simulations and machine learning to accelerate food R&D
- Practical examples and case studies that impact food product development
- To explore key areas in your R&D where physics-based simulation and machine learning can provide value

Dr. Jeffrey Sanders
Product Manager
Jeff Sanders received his B.S. in applied physics from Worcester Polytechnic Institute and then his Ph.D. in biophysics and molecular pharmacology from Thomas Jefferson Medical College. Since joining Schrödinger in 2013, he has served several roles and is currently the product manager and scientific lead for the consumer packaged goods applications group. Additionally, he is a managing board member of the Food Engineering, Expansion, and Development (FEED) institute and holds an adjunct position in the department of food science at University of Massachusetts, Amherst.
Overview
With the rise in utility and access to artificial intelligence (AI) solutions in everyday life, the food industry is searching for practical use cases to leverage its power. While some claim AI will render traditional research and development in the food industry obsolete, the paradigm shift has yet to come to fruition. In order for a digital transformation of such scale to occur, data will become the key driver.
In food science, data collection is often sparse, or is collected at the macroscopic scale with little insight to the underlying physical and chemical driving forces. Unlike AI (also called machine learning), physics-based simulation is able to generate data based on realistic computational models of food products, processing, and packaging materials. The data generated is interpretable, allowing researchers and engineers to make informed decisions before embarking on costly experimental testing. By leveraging data generated from physics-based simulations at the molecular level combined with existing experimental data where available, machine learning models can then be generated overcoming the data sparsity issue often encountered. More importantly, physics-based simulations can help researchers develop models that are both interpretable and testable.
In this talk, we will explore how physics-based simulations are used in food research and the synergy that can be achieved when they are combined with machine learning models.
What our alumni say