
- Publication
- Jan 14, 2003
Binding site characteristics in structure-based virtual screening: evaluation of current docking tools
Schulz-Gasch, et al. J. Mol. Mod., 2003, 9, 47-57
- Publication
- Jan 9, 2003
A Computationally Inexpensive Modification of the Point Dipole Electrostatic Polarization Model for Molecular Simulations
Kaminski, et al. J. Comput. Chem., 2003, 24, 267-276
- Publication
- Jan 8, 2003
Stereostructure-Activity Studies on Agonists at the AMPA and Kainate Subtypes of Ionotropic Glutamate Receptors
Johansen, et al. Chirality, 2003, 15, 167-179
- Publication
- Jan 6, 2003
Design of Novel N-(2,4-Dioxo-1,2,3,4-tetrahydro-thieno[3,2-d]pyrimidin-7-yl)-guanidines as Thymidine Phosphorylase Inhibitors, and Flexible Docking to a Homology Model
Price, et al. Bioorganic & Medicinal Chem. Lett., 2003, 13, 107-110
- Publication
- Nov 23, 2002
Metal-Dependent Inhibition of HIV-1 Integrase
Neamati, et al. J. Med. Chem., 2002, 45, 5661-5670
- Publication
- Nov 15, 2002
Hydroxylation of Methane by Non-Heme Diiron Enzymes: Molecular Orbital Analysis of the C-H Bond Activation by Reactive Intermediate Q
Baik, et al. J. Am. Chem. Soc., 2002, 124, 14608-14615
- Publication
- Nov 13, 2002
Chemosensors for the Marine Toxin Saxitoxin
Gawley, et al. J. Am. Chem. Soc., 2002, 124, 13448-13453
- Publication
- Oct 15, 2002
Development of a Polarizable Force Field for Proteins via ab initio Quantum Chemistry: First Generation Model and Gas Phase Tests
Kaminski, et al. J. Comput. Chem., 2002, 23, 1515-1531
- Publication
- Oct 15, 2002
Force Field Validation Using Protein Side Chain Prediction
Jacobson, et al. J. Phys. Chem. B., 2002, 106, 11673-11680
- Publication
- Oct 7, 2002
Molecular Mechanics Calculations as Predictors of Enantioselectivity for Chiral Nucleophile Catalyzed Reactions
Taggi, et al. Tetrahedron, 2002, 58, 8351-8356
- Publication
- Aug 13, 2002
Selective Agonists at Group II Metabotropic Glutamate Receptors: Synthesis, Stereochemistry, and Molecular Pharmacology of (S)- and (R)-2-Amino-4-(4-hydroxy[1,2,5]thiadiazol-3-yl) butyric Acid
lausen, et al. J. Med. Chem., 2002, 45, 4240-4245
- Publication
- Jul 18, 2002
Computing Redox Potentials in Solution: Density Functional Theory as a Tool for Rational Design of Redox Agents
Baik, et al. J. Phys. Chem. A, 2002, 106, 7407-7412
Events
 Event
Life Science
Event
Life Science
- Jul 6th-9th, 2026
CRS 2026
Schrödinger is excited to be participating in the CRS 2026 conference taking place on July 6th – 9th in Lisbon, Portugal.
 Event
Life Science
Event
Life Science
- Jul 9, 2026
Lunch and Learn: Biotherapeutics Design: Physics-Informed AI for Protein Engineering
Join Schrödinger scientists for a Lunch & Learn on the future of antibody/antigen engineering.
 Event
Materials Science
Event
Materials Science
- Jul 12th-15th, 2026
IFT FIRST 2026
Schrödinger is excited to be participating in the IFT First Annual Event and Expo 2026 conference taking place on July 12th – 15th in Chicago, Illinois.
Webinars
 Webinar
Life Science
Webinar
Life Science
- May 27, 2026
Biologics modeling for wet lab scientists: Detecting and deprioritizing dead ends before they reach the bench recording
Join us to learn how to detect and deprioritize high-risk candidates, effectively discarding developability dead-ends before they ever reach the bench.
 Webinar
Life Science
Webinar
Life Science
- Apr 30, 2026
Educator’s Week 2026
Join us for a series of live webinar presentations from leading educators at top academic institutions, as well as talks by Schrödinger scientists.
 Webinar
Life Science
Webinar
Life Science
- Apr 24, 2026
Schrödinger デジタル創薬セミナー ~計算化学がもたらす創薬プロセスの変貌~ 第24回
APR 24, 2026 | Diverse computational strategies enable the discovery of p38α-MK2 molecular glues
Documentation
- Documentation
Learning Path: Oligonucleotide Modeling
A structured overview of tools and workflows for nucleic acids in drug discovery.
- Documentation
WaterMap
Efficiently converged MD simulations are run with explicit water molecules, and resultant trajectories are analyzed to cluster hydration sites.
- Documentation
SiteMap
Identify binding sites, including allosteric binding sites and protein-protein interfaces, and evaluate their druggability.
Tutorials
- Tutorial
Structure-Based Virtual Screening using Glide
Prepare receptor grids for docking, dock molecules and examine the docked poses.
- Tutorial
Ligand Binding Pose Generation for FEP+
Generate starting poses for FEP simulations for a series of BACE1 inhibitors using core constrained docking.
- Tutorial
Homology Modeling of Protein-Ligand Binding Sites with IFD-MD
Create a homology model of TYK2 from JAK3 and including a bound ligand. Compare this model with the crystal structure for TYK2 bound to 4GIH.
Training Videos
 Video
Life Science
Video
Life Science
Getting Going with Maestro BioLuminate
A free video series introducing the basics of using Maestro Bioluminate.
 Video
Life Science
Video
Life Science
- Video
Introducing Ligand Designer
An overview of the LigandDesigner workflow, Editing in 2D and 3D, using display options and overlays, and accessing the Admin Panel.
Publications
- Publication
- May 8, 2026
Discovery of 2H-Pyrrolo[3,4-c]pyridin-3-one Derivatives as Type-III c-MET Inhibitors Enabled by Free-Energy Perturbation CalculationsCl
Therrien, et al. ACS Medicinal Chemistry Letters, 2026
- Publication
- Apr 8, 2026
Structure-Based Discovery of Imidazo[4,5-c]pyridine SARM1 Modulators Showing Paradoxical Activation
Albanese, et al. Journal of Medicinal Chemistry, 2026, 69(8), 9521–9536
- Publication
- Mar 21, 2026
Structure-Based Calculation of Excipient Effects on the Viscosity of Concentrated Antibody Solutions
Shelley, et al. mAbs, 2026, 18(1)
Case Studies
 Case Study
Life Science
Materials Science
Case Study
Life Science
Materials Science
 Case Study
Life Science
Case Study
Life Science
 Case Study
Life Science
Case Study
Life Science
White Papers
 White Paper
Life Science
White Paper
Life Science
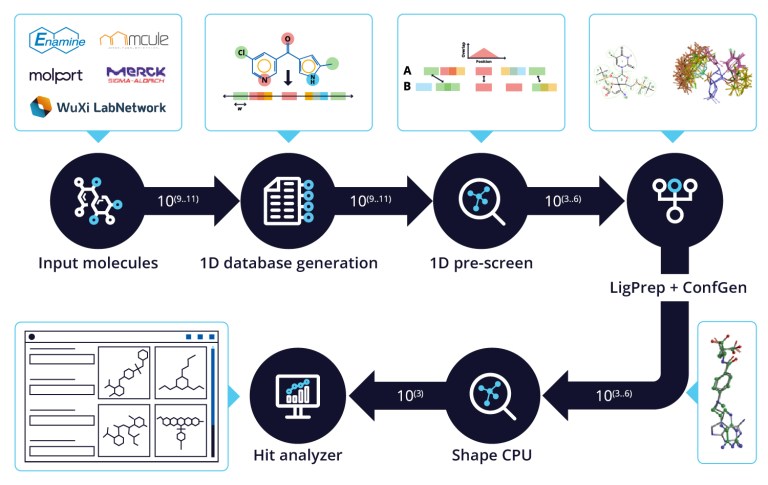
- Jan 29, 2026
FEP+ Pose Builder — maximizing utility and productivity in FEP simulations
FEP+ Pose Builder is a methodological advancement introduced as an integrated feature to drastically enhance accessibility, user-friendliness, and productivity within the FEP+ pipeline.
 White Paper
Life Science
White Paper
Life Science
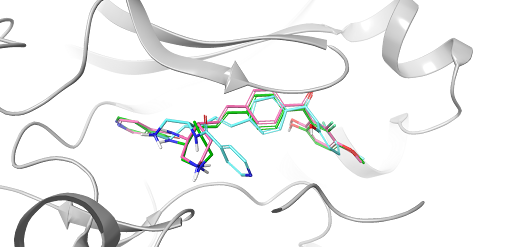
- Oct 29, 2024
20 Years of Glide: A Legacy of Docking Innovation and the Next Frontier with Glide WS
Glide has long set the gold standard for commercial molecular docking software due to its robust performance in both binding mode prediction and empirical scoring tasks, ease of use, and tight integration with Schrödinger’s Maestro interface and molecular discovery workflows.
 White Paper
Life Science
White Paper
Life Science
Quick Reference Sheets
- Quick Reference Sheet
Force Field Builder
A one-page guide to calculate missing torsion parameters for ligands using the Force Field Builder panel.
- Quick Reference Sheet
Ligand Interaction Diagram
A one-page guide to using the Ligand Interaction Diagram for examining ligand-receptor interactions.
- Quick Reference Sheet
GlideMap
A one-page guide to using the GlideMap GUI for ligand placement guided by experimental density.

Latest insights from Extrapolations blog

With FEP+, “The Experiment is the Limit.”
Over the past century, small molecule drugs have represented the dominant modality in drug research, enabling medical breakthroughs that have saved countless lives.

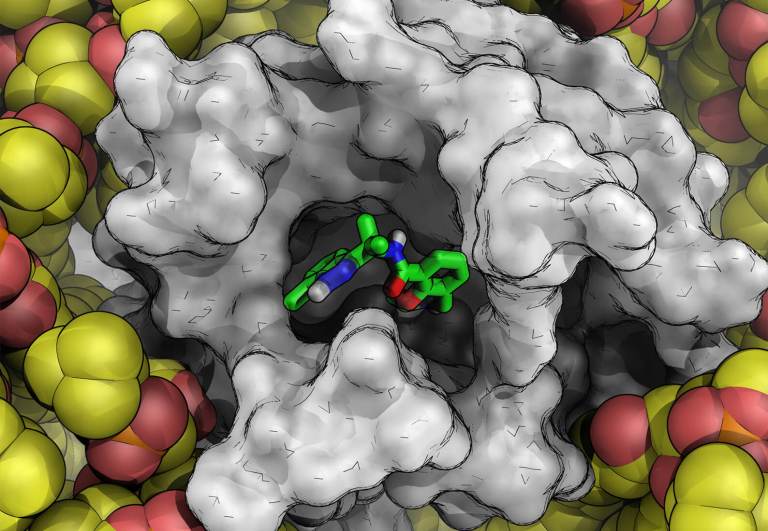
Tackling Drug Solubility: AbbVie and Schrödinger Collaborate to Advance Accurate Prediction Methods
The complexity and size of drug candidates has grown in recent years as scientists pursue novel targets once considered undruggable.
 Blog
Blog
Can AlphaFold Models be Used for Structure-Based Drug Design? A Perspective Two Years In
We recently sat down with Edward Miller, Senior Director of Protein Structure Modeling at Schrödinger, to discuss his experience using AlphaFold models for SBDD.
Training & Resources
Online certification courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Free learning resources
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Other Resources