
- Publication
- Jul 12, 2002
On the Role of Crystal Packing Forces in Determining Protein Sidechain Conformations
Jacobson, et al. J. Mol. Biol., 2002, 320, 597-608
- Publication
- Jun 26, 2002
Toward Computing Relative Configurations: 16-epi-Latrunculin B, a New Stereoisomer of the Actin Polymerization Inhibitor Latrunculin B
Hoye, et al. J. Am. Chem. Soc., 2002, 124, 7405-7410
- Publication
- Jun 1, 2002
Complete Protein Structure Determination Using Backbone Residual Dipolar Couplings and Sidechain Rotamer Predication
Andrec, et al. J. of Structural and Functional Genomics, 2002, 2, 103-111
- Publication
- Mar 31, 2002
Prediction of Drug Solubility from Structure
Jorgensen, et al. Adv. Drug Deliv. Rev., 2002, 54, 355-366
- Publication
- Jan 30, 2002
Application of the Frozen Atom Approximation to the GB/SA Continuum Model for Solvation Free Energy
Guvench, et al. J. Comput. Chem., 2002, 23, 214-221
- Publication
- Jan 25, 2002
Accurate Prediction of Acidity Constants in Aqueous Solution via Density Functional Theory and Self-Consistent Reaction Field Methods
Klicic, et al. J. Phys. Chem. A, 2002, 106, 1327-1335
- Publication
- Dec 19, 2001
Fully Flexible Low-Mode Docking: Application to Induced Fit in HIV Integrase
Keseru, et al. J. Am. Chem. Soc., 2001, 123, 12708-12709
- Publication
- Dec 1, 2001
Reproducing the Conformations of Protein-bound Ligands: A Critical Evaluation of Several Popular Conformational Searching Tools
Bostrom, et al. J. Comput.-Aided Mol. Des., 2001, 15, 1137-1152
- Publication
- Nov 22, 2001
Novel Agouti-Related-Protein-Based Melanocortin-1 Receptor Antagonist
Thirumoorthy, et al. J. Med. Chem., 2001, 44, 4114-4124
- Publication
- Sep 21, 2001
New Linear Interaction Method for Binding Affinity Calculations Using a Continuum Solvent Model
Zhou, et al. J. Phys. Chem. B, 2001, 105, 10388-10397
- Publication
- May 1, 2001
A Comparison of Methods for Pharmacophore Generation with the Catalyst Software and their Use for 3D-QSAR: Application to a Set of 4-aminopyridine Thrombin Inhibitors
Greenidge, et al. Mini Rev. Med. Chem., 2001, 1, 79-87
- Publication
- Mar 28, 2001
Activation of the C-H bond of Methane by Intermediate Q of Methane Monoozygenase: A Theoretical Study
Gherman, et al. J. Am. Chem. Soc., 2001, 123, 3836-3837
Events
 Event
Materials Science
Event
Materials Science
- Aug 11th-18th, 2026
IUCr2026
Schrödinger is excited to be participating in the IUCr2026 conference taking place on August 11th – 18th in Calgary, Alberta, Canada.
 Event
Materials Science
Event
Materials Science
- Aug 18th-21st, 2026
IMID 2026
Schrödinger is excited to be participating in the 26th International Meeting on Information Display conference taking place on August 18th – 21st in Busan, South Korea.
 Event
Life Science
Event
Life Science
- Sep 6th-10th, 2026
EFMC Medicinal Chemistry 2026
Schrödinger is excited to be participating in the EFMC Medicinal Chemistry 2026 conference taking place on September 6th – 10th in Basel, Switzerland.
Webinars
 Webinar
Life Science
Webinar
Life Science
- May 27, 2026
Biologics modeling for wet lab scientists: Detecting and deprioritizing dead ends before they reach the bench recording
Join us to learn how to detect and deprioritize high-risk candidates, effectively discarding developability dead-ends before they ever reach the bench.
 Webinar
Life Science
Webinar
Life Science
- May 13, 2026
Inside the design loop: A day in the life of a digital medicinal chemist recording gated
In this session, we’re stepping away from the high-level overviews to give you an honest look at how medicinal chemists at Schrödinger actually work.
 Webinar
Life Science
Webinar
Life Science
- Apr 30, 2026
Educator’s Week 2026
Join us for a series of live webinar presentations from leading educators at top academic institutions, as well as talks by Schrödinger scientists.
Documentation
- Documentation
Learning Path: Oligonucleotide Modeling
A structured overview of tools and workflows for nucleic acids in drug discovery.
- Documentation
WaterMap
Efficiently converged MD simulations are run with explicit water molecules, and resultant trajectories are analyzed to cluster hydration sites.
- Documentation
SiteMap
Identify binding sites, including allosteric binding sites and protein-protein interfaces, and evaluate their druggability.
Tutorials
- Tutorial
Structure-Based Virtual Screening using Glide
Prepare receptor grids for docking, dock molecules and examine the docked poses.
- Tutorial
Ligand Binding Pose Generation for FEP+
Generate starting poses for FEP simulations for a series of BACE1 inhibitors using core constrained docking.
- Tutorial
Homology Modeling of Protein-Ligand Binding Sites with IFD-MD
Create a homology model of TYK2 from JAK3 and including a bound ligand. Compare this model with the crystal structure for TYK2 bound to 4GIH.
Training Videos
 Video
Life Science
Video
Life Science
Getting Going with Maestro BioLuminate
A free video series introducing the basics of using Maestro Bioluminate.
 Video
Life Science
Video
Life Science
- Tutorial
- Video
The LiveDesign Assistant
Learn how to build and adjust coloring rules, create Freeform and Formula columns, and plot data using the LiveDesign Assistant.
Publications
- Publication
- May 8, 2026
Discovery of 2H-Pyrrolo[3,4-c]pyridin-3-one Derivatives as Type-III c-MET Inhibitors Enabled by Free-Energy Perturbation CalculationsCl
Therrien, et al. ACS Medicinal Chemistry Letters, 2026
- Publication
- Apr 8, 2026
Structure-Based Discovery of Imidazo[4,5-c]pyridine SARM1 Modulators Showing Paradoxical Activation
Albanese, et al. Journal of Medicinal Chemistry, 2026, 69(8), 9521–9536
- Publication
- Mar 21, 2026
Structure-Based Calculation of Excipient Effects on the Viscosity of Concentrated Antibody Solutions
Shelley, et al. mAbs, 2026, 18(1)
Case Studies
 Case Study
Life Science
Materials Science
Case Study
Life Science
Materials Science
 Case Study
Life Science
Case Study
Life Science
 Case Study
Life Science
Case Study
Life Science
White Papers
 White Paper
Life Science
White Paper
Life Science
- Jan 29, 2026
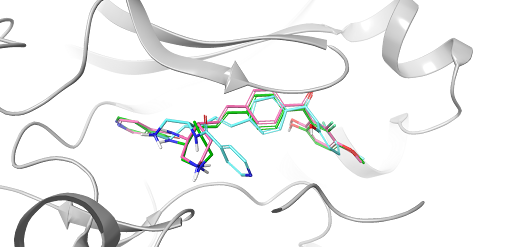
FEP+ Pose Builder — maximizing utility and productivity in FEP simulations
FEP+ Pose Builder is a methodological advancement introduced as an integrated feature to drastically enhance accessibility, user-friendliness, and productivity within the FEP+ pipeline.
 White Paper
Life Science
White Paper
Life Science
- Oct 29, 2024
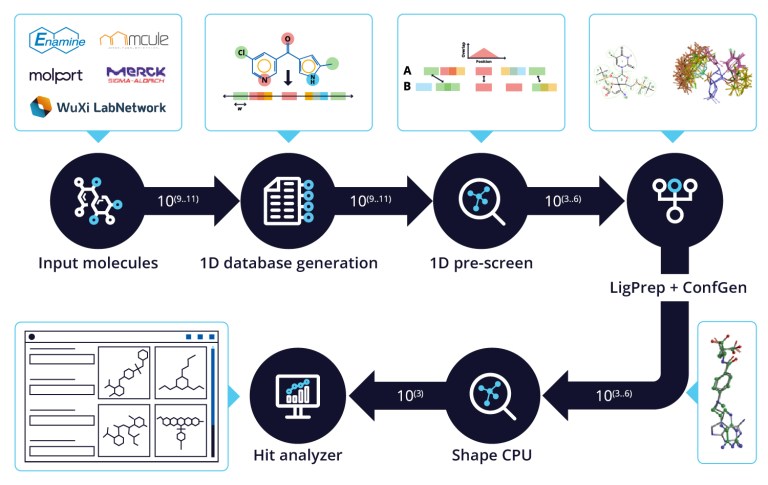
20 Years of Glide: A Legacy of Docking Innovation and the Next Frontier with Glide WS
Glide has long set the gold standard for commercial molecular docking software due to its robust performance in both binding mode prediction and empirical scoring tasks, ease of use, and tight integration with Schrödinger’s Maestro interface and molecular discovery workflows.
 White Paper
Life Science
White Paper
Life Science
Quick Reference Sheets
- Quick Reference Sheet
Force Field Builder
A one-page guide to calculate missing torsion parameters for ligands using the Force Field Builder panel.
- Quick Reference Sheet
Ligand Interaction Diagram
A one-page guide to using the Ligand Interaction Diagram for examining ligand-receptor interactions.
- Quick Reference Sheet
GlideMap
A one-page guide to using the GlideMap GUI for ligand placement guided by experimental density.

Latest insights from Extrapolations blog

With FEP+, “The Experiment is the Limit.”
Over the past century, small molecule drugs have represented the dominant modality in drug research, enabling medical breakthroughs that have saved countless lives.

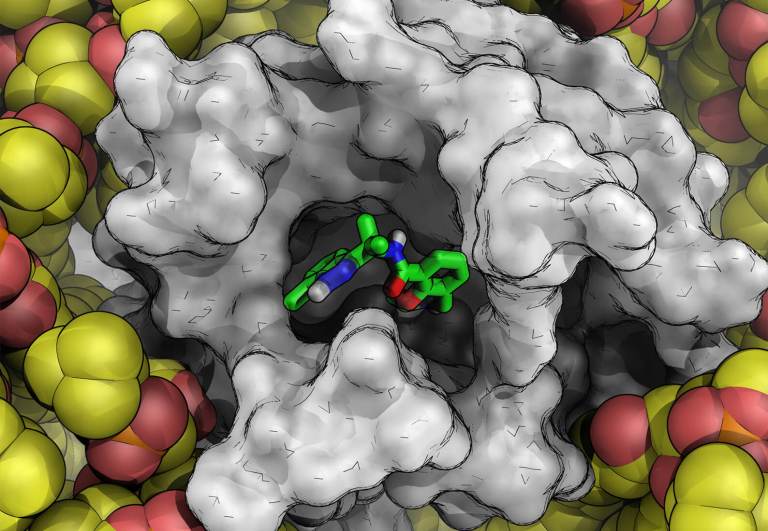
Tackling Drug Solubility: AbbVie and Schrödinger Collaborate to Advance Accurate Prediction Methods
The complexity and size of drug candidates has grown in recent years as scientists pursue novel targets once considered undruggable.
 Blog
Blog
Can AlphaFold Models be Used for Structure-Based Drug Design? A Perspective Two Years In
We recently sat down with Edward Miller, Senior Director of Protein Structure Modeling at Schrödinger, to discuss his experience using AlphaFold models for SBDD.
Training & Resources
Online certification courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Free learning resources
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Other Resources