As artificial intelligence (AI) and machine learning (ML) technologies rapidly advance, materials scientists, executives, and R&D professionals are being tasked with developing an AI/ML strategy to drive innovation. Yet while AI/ML tools may advertise the appeal of push-button innovation, this is almost never the reality. Even the term AI itself can be a misleading buzzword.
Schrödinger is uniquely positioned to partner with materials R&D teams to execute AI/ML strategies that deliver true business value because we leverage the proven accuracy of physics-based modeling, the speed and scale of machine learning, and the deep domain expertise of our materials scientists. So while AI/ML technologies can be transformative, gaining a competitive advantage with AI is only possible with the talent, vision, and know-how of people who can utilize and direct these technologies towards meaningful, impactful outcomes.
Personalized human support is critical for project efficiency and productivity
Schrödinger’s professional software support is unique in providing domain expertise, personalized assistance, and reliable service. This human difference ensures that users can fully leverage the potential of digital tools to address complex issues through tailored guidance and support.
Schrödinger Support
Benefits
24-7 global support from a team of experts
Faster project timelines
On-site and virtual assistance with expert application scientists
Technical discussions, troubleshooting, and knowledge transfer
Extensive online training, tutorials, and resources
Skilled users to maximize outcomes
Expert-led customized modeling service packages
Novel state-of-the-art research, knowledge transfer and improved success rates
“As a former industrial modeler at Boeing, I understand that the needs of each client are unique. That’s why we focus on delivering personalized support that addresses their specific challenges in R&D digitization.”
Andrea BrowningDirector of Polymers and Soft Matter
Schrödinger technical experts increase capacity and capability
Through strategic partnerships or customized contract research, Schrödinger’s team of expert scientists work closely with customers to tackle challenging problems by deploying digital chemistry strategies to guide rapid materials design and optimization. By working as a team, we share the same challenges and goals. These expert-driven collaborations transform challenges into opportunities, driving innovation and delivering exceptional results.
“Over the course of 20 years working with companies in this sector, I am convinced that collaboration is the key to delivering value, by listening to industry needs and tailoring the modeling solution.”
Simon ElliottDirector of Atomic Level Process Simulation
Expertise and industry-grade software can be the difference between project success and failure
It is common for materials science researchers to piece together a variety of modeling and simulation tools. Free software packages are often limited by outdated documentation, insufficient support, and obstacles to integration and automation. The human element is one of the key differentiators between industry-grade software like Schrödinger and less sophisticated software packages.
Schrödinger has a large team working on user experience, technical support, and education. This team ensures that clients can seamlessly access the science underpinning the simulations and use it at a scale that allows them to get their job done.
“Having used free software extensively in the past, I now realize how much time I was spending on troubleshooting rather than actual research. Schrödinger’s solutions are a game-changer for productivity.”
Pavel DubSenior Principal Scientist and Product Manager
Advanced physics-based modeling and AI/ML software for R&D teams
Schrödinger brings more than 30 years of scientific innovation and deep domain expertise to empower R&D teams to solve their unique challenges with computational approaches.
“By working closely with Schrödinger experts, we were impressed by how fast we were able to learn to apply molecular simulations, even with no prior modeling experience. Our collaborations have been very successful, not only because of our satisfaction with Schrödinger’s advanced technologies, but also because of their level of scientific expertise, support, and collaborative openness.”
Martin SettleSenior Research Manager, Reckitt
The resin design and incubation team at SABIC worked closely with Schrödinger’s material science team to build accurate machine learning (ML) models to speed up the discovery of new polymers. “These computational results are highly promising and can potentially shorten our polymer innovation timelines from traditionally a couple of years to only a couple of months.”
Vaidya RamakrishnanStaff Scientist, SABIC
“Schrödinger provides us with more than just software as part of our service agreement—they are a true partner in our research. With an office here in Japan, Schrödinger scientists and engineers are easily accessible and able to collaborate in-person with our team.”
Nobuyuki N. MatsuzawaExecutive Engineer, Panasonic Industry Co., Ltd.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Complex chemical mixtures — or formulations — are used in a wide range of applications, such as gasoline blends in oil & gas, daily care products in consumer goods, and drug delivery in pharmaceutics. Given the vast number of potential formulations, evolving regulatory requirements, and increasing consumer demand for eco-friendly and sustainable products, we need innovative and cost-effective solutions for designing enhanced formulations. The latest advancements in atomic-scale modeling and machine learning (ML) have enabled computer-aided screening of large numbers of formulation candidates — thus, accelerating the identification of promising formulations and reducing costly experiments.
Schrödinger’s Formulation Machine Learning tool uses data-driven methods to correlate ingredient structure and composition to formulation properties. This tool uses advanced cheminformatics descriptors and automatic hyperparameter tuning to find the best ML model, and allows external features (e.g., temperature, pressure) from experiments or high-throughput molecular dynamics (MD) calculations to be used as additional input to the ML model. The Formulation ML tool enables R&D teams to quickly train and deploy ML models to rapidly explore the broad design space of formulations by varying the chemical ingredients, compositions, and external features.
Advantages of Schrödinger Formulation Screening Technology
Efficient ML model building and data generation: Leveraging deep learning technology to build accurate ML models to predict formulation properties, which can be coupled with MD simulations as a way to generate physically meaningful descriptors to improve ML model accuracy
Scalable: ML can be trained and evaluated for mixtures with more than 100 components, extending the capabilities beyond simple mixtures to designing complex mixtures with enhanced properties
Automated: Automatic hyperparameter tuning enables accurate ML model development using expert cheminformatic descriptors with minimal ML expertise required
Rapid screening capabilities: ML can generate predictions in a fraction of a second, which can scale up to screening ~100K formulations in the order of minutes-hours
Dedicated support: Dedicated support team consisting of scientific experts at Schrödinger are available to help users apply computational tools to their applications
Multiple platform functionality: Can be used on laptops, desktops, and high performance clusters
Applications Across Industries
Consumer Products
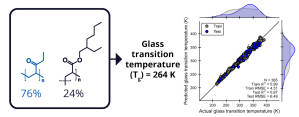
Random copolymer systems are often found in packaging materials, and glass transition temperature (Tg) is an important parameter that dictates the stability of the polymer as a function of temperature. Formulation ML can accurately predict Tg for 365 examples with a test set coefficient of determination (R2) of 0.97.2
Energy Storage
Liquid electrolytes are often used in batteries to facilitate the movement of electrical charge between an anode and cathode, and viscosity is an important parameter that dictates how easy ions can move through an electrolyte solution. Formulation ML can accurately predict temperature-dependent viscosity given ~34K examples with a test set R2 of 0.96.3
Pharmaceutical Formulation
Solubility of drug molecules in pure and binary mixture solutions is crucial for drug delivery applications for pharmaceutical formulations. Formulation ML can accurately predict temperature-dependent drug solubility for either pure or binary mixture solutions given ~27K examples, which achieves a test set R2 of 0.93.4
Oil and Gas
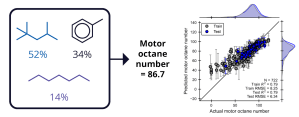
Mixtures of hydrocarbons are critical in gasoline blends, facilitating efficient combustion for automotive engines, and motor octane number (MON) is an important parameter that measures the fuel behavior under external pressure. Formulation ML can accurately predict MON given ~700 examples with the number of components ranging from pure (single) component systems to 120 components, which achieves a test set R2 of 0.79.5
Learn more in the tutorial (Note: you will need a web account to access tutorials)
References
Leveraging high-throughput molecular simulations and machine learning for the design of chemical mixtures
Alex, C., et al. npj Comput Mater 11, 72, 2025,https://doi.org/10.1038/s41524-025-01552-2.
The glass transition temperature of random copolymers: 1. Experimental data and the Gordon-Taylor equation
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Join us on Monday, October 14th at Basel Marriott Hotel for an immersive workshop on the application of physics-based computational methods (BioLuminate, FEP+) and collaborative enterprise informatics (LiveDesign) towards designing better biologics, faster. We welcome those who are new to computational tools or those who already have some experience and seek a more structured understanding, including of new and advanced methods. During the session, enjoy a networking lunch to connect with fellow attendees.
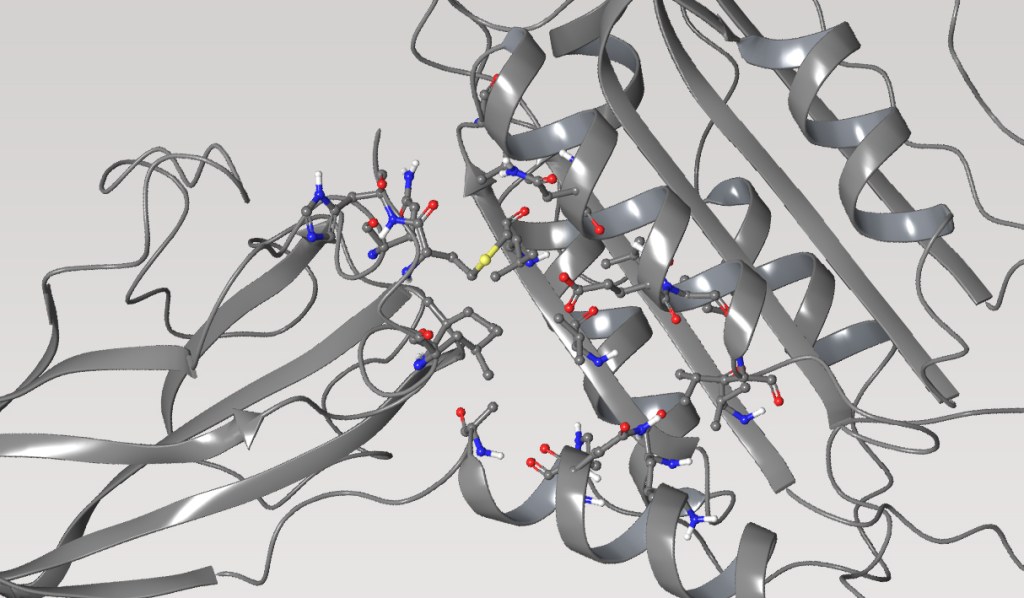
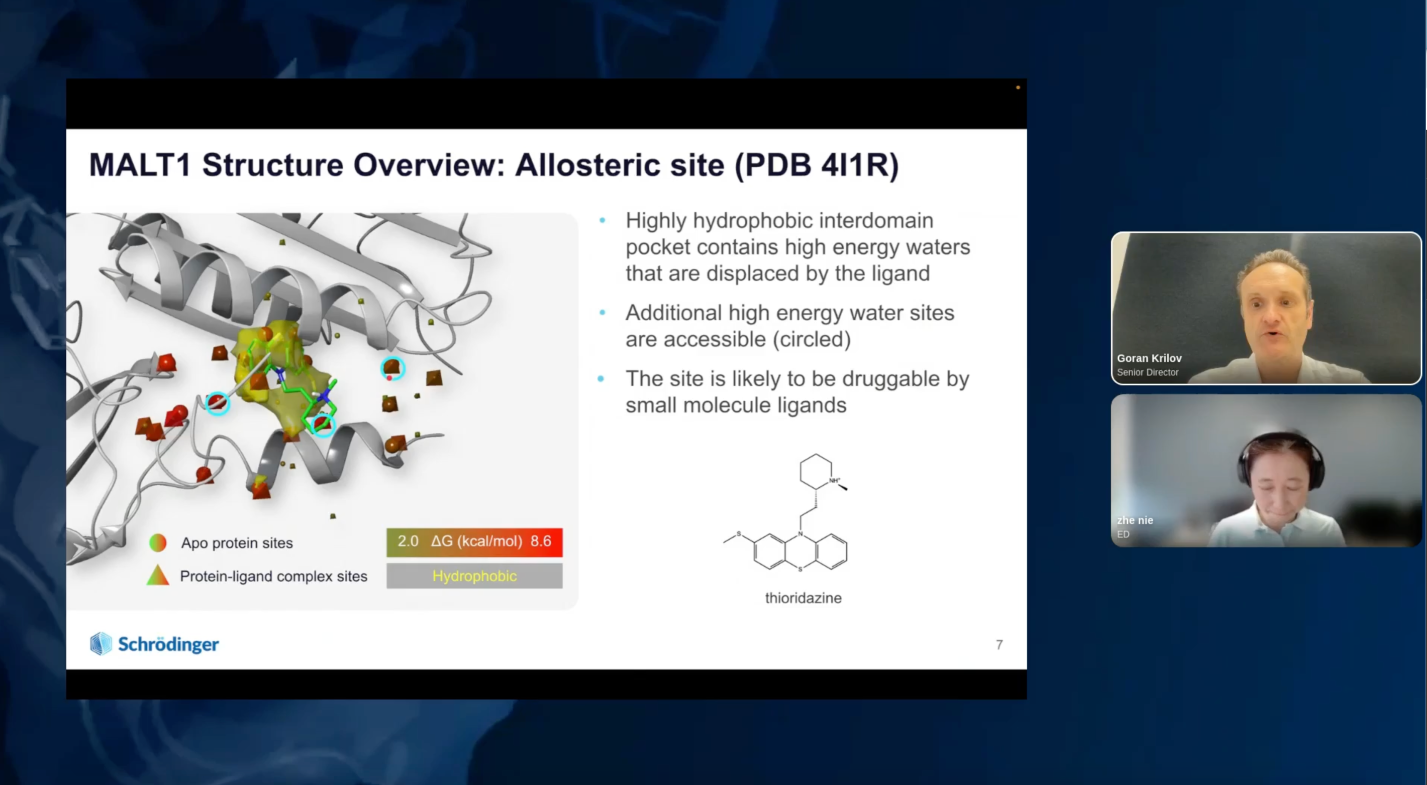
Accelerated in silico discovery of SGR-1505: A potent MALT1 allosteric inhibitor for the treatment of mature B-cell malignancies
Share
Abstract:
MALT1 (Mucosa-associated lymphoid tissue lymphoma translocation protein 1) is a component of the MALT1-BCL10-CARD11 complex downstream from the Bruton Tyrosine Kinase (BTK) on the B-cell receptor signaling pathway. MALT1 is a key mediator of nuclear factor kappa B (NF-κB) signaling, which is the main driver of a subset of B-cell lymphomas. MALT1 is considered a potential therapeutic target for several subtypes of non-Hodgkin’s B-cell lymphomas and chronic lymphocytic leukemia (CLL), including tumors with acquired BTK inhibitor (BTKi) resistance. Constitutive activation of the NF-κB is a molecular hallmark of activated B cell-like diffuse large B cell lymphoma (ABC-DLBCL), and MALT1 may have utility as a treatment option for ABC-DLBCL. Furthermore, a third-party MALT1 inhibitor recently showed strong anti-tumor activity in mature B cell malignancies from Phase 1 studies.
By applying advanced physics-based modeling techniques, including combining free energy calculations with machine learning methods and chemistry-aware compound enumeration workflow, the Schrödinger team explored extensive sets of de novo design ideas to quickly identify a novel hit series with an in vivo tool molecule to establish an in vivo PD and efficacy mouse model early on in the project. Multi-parameter optimization (MPO) allowed efficient prioritization of molecules with good potency and drug-like properties during lead optimization. This led to the discovery of a highly potent MALT1 inhibitor, SGR-1505, with a well-balanced property profile in under a year, with only 78 compounds synthesized in the lead series and 129 compounds overall. SGR-1505 is a potent and orally available allosteric MALT1 inhibitor. It demonstrated strong anti-tumor activity alone and in combination with BTK inhibitors in multiple in vivo B-cell lymphoma xenograft models. Currently, a Phase 1 clinical trial with SGR-1505 in patients with mature B-cell neoplasms is ongoing (NCT05544019).
Webinar Highlights:
Discover how free energy calculations, amplified by machine learning methods, led to the discovery of a highly potent MALT1 inhibitor, SGR-1505
Learn how the team used an MPO scoring function consisting of FEP+-based predictions of affinity and solubility, physics-based predictions of permeability, and predictions of lipophilicity to optimize compounds
Ask questions to gain further insight from the speakers to apply to your work
Our Speakers
Goran Krilov
Senior Director, Schrödinger
Dr. Goran Krilov is a senior Director of Computational Chemistry at Schrödinger’s Therapeutic Group. For the past twenty five years, his work has focused on developing and applying cutting-edge computational chemistry techniques to problems in biophysics and drug discovery. He has led the modeling efforts on a number of internal projects as well as external collaborations in oncology and neurogenerative diseases, resulting in two clinical candidates currently undergoing Phase I trials. Prior to joining Schrödinger, Dr. Krilov has worked in both industry and academia, including IBM, Boston College snd Strand Life Sciences.
Zhe Nie
Executive Director, Schrödinger
Dr. Zhe Nie is the Executive Director of Medicinal Chemistry at Schrödinger’s Therapeutic Group. She has been leading multiple wholly owned and partnered drug discovery programs at Schrödinger. Most recently, she led Schrödinger’s MALT1 discovery project team, successfully developed the small molecule drug SGR-1505 (Schrödinger’s first internal clinical asset currently in Ph1) applying Schrödinger’s computational platform. It took less than two years from the start of the project to the selection of the clinical candidate. She also led the DLK collaboration project with Takeda Pharmaceuticals which discovered a potent, selective, and brain-penetrate DLK inhibitor as a promising preclinical candidate for the treatment of neurodegenerative diseases using Schrödinger’s computational platform. She has extensive experiences in applying advanced computational tools to assist in the design of small molecule drug candidates. She previously worked at Takeda, Celgene and Quanticel Pharmaceuticals (acquired by Celgene), led and contributed to advancing multiple small molecule drugs to the clinics including TAK-960, TAK-659 and CC-90011.
Molecular modeling is a powerful computational technique widely used in materials science to predict and understand the properties and behavior of materials at the molecular level. By simulating the interactions between atoms and molecules, researchers can explore the structural, mechanical, electronic, and thermal properties of various materials and gain a deeper understanding. Molecular modeling encompasses a range of methods, including molecular dynamics, quantum mechanics, and coarse grained simulations, each providing unique insights into material properties and guiding experimental efforts.
The integration of molecular modeling into materials science can accelerate the development of advanced materials for applications in pharmaceuticals, Fast Moving Consumer Goods, electronics, energy storage, catalysis, and more.
This webinar series “From molecules to Materials Applications” will delve into molecular modeling techniques and their transformative impact on Materials Science research using the Schrödinger Materials Science tools.
Molecular Modeling: A Key to Solving Real-Life Challenges in Pharma Formulations
Speaker: Sudharsan Pandiyan, Principal Scientist II, Schrödinger
Abstract: The demand for innovative drug delivery methods has driven researchers to explore the intricate structure-property relationships within pharmaceutical formulations. Quantum Mechanical (QM) and Molecular Dynamics (MD) simulations are powerful tools for understanding these formulations at a molecular level.
Key areas of interest in pharmaceutical sciences include chemical stability, reactivity, molecular degradation, impurity profiling, excipient selection, and polymorph prediction. A thorough understanding of the Active Pharmaceutical Ingredient (API) is essential before embarking on the formulation development process.
The Schrödinger Materials Science Suite (MS-Suite) offers comprehensive computational workflows to predict spectra (IR, Raman, NMR, UV-Visible, XRD) and assess the API’s behavior under varying pH conditions, including its degradation pathways and chemical reactivity.
Recent advancements in GPU technology have significantly accelerated MD simulations, enabling previously unattainable time scales. This dramatic speedup, combined with predictive accuracy, is poised to revolutionize the use of MD simulations in pharmaceutical formulation development. MD-based workflows can help us address critical formulation design questions on physical stability of formulations, phase transitions, miscibility, solubility and diffusion of API through membranes, morphology, excipient compatibility, encapsulation, and coating selections.
This presentation will highlight several successful case studies that demonstrate these capabilities from a molecular perspective.
SEPT 18 | 18:00 IST
Harnessing Molecular Modeling to transform innovation in Polymeric Materials and Consumer Packaged Goods
Speaker: Sriram Krishnamurthy, Senior Scientist I, Schrödinger
Abstract: Polymeric materials and consumer packaged goods (CPGs) hold significant importance in both industrial and everyday contexts, impacting numerous aspects of modern life. Polymers play a crucial role in various industries due to their versatility and wide range of applications like construction, electronics, healthcare, and automotive. As scientific research and innovation continue to advance, polymeric materials remain at the forefront of development. In the context of consumer packaged goods (CPG), which significantly influence our daily lives, there is an ongoing push to create products that meet evolving consumer demands, such as healthier food options and more sustainable packaging solutions. Polymeric materials and CPGs are deeply interconnected in their importance to modern society.
Molecular modeling has emerged as a transformative tool in the design and optimization of these materials. By providing deep insights into molecular interactions and material properties, molecular modeling accelerates the development of novel, efficient, and sustainable materials. This approach not only enhances our understanding of material behavior, but also facilitates the innovation of advanced solutions tailored to the specific needs of modern consumer products.
This webinar will highlight Schrödinger’s Materials Science tools that can accelerate R&D efforts in these scientific domains. We will showcase practical case studies to tackle key problems and identify areas where molecular modeling can be applied.
SEPT 25 | 18:00 IST
Efficient Computation of Process Parameters for Controlling the Chemistry of Deposition or Etch
Speaker: Simon Elliott, Research Leader, Schrödinger
Abstract: We present a variety of computational techniques for understanding, controlling and improving deposition and etch processes. The emphasis is on choosing the right technique for the research question and time available. The same computational techniques can be used to investigate other gas-surface processes, such as catalysis or sensing.
Different chemical processes can be in competition when a solid surface is treated with a gaseous reagent and the outcome is determined by conditions such as temperature and pressure. For instance, continuous deposition (CVD) may take over from self-limiting deposition (ALD) as the temperature is raised. Or temperature may dictate which material is deposited; in the case presented here, ruthenium oxide film is deposited from RuO4+H2 in experiments at 75°C, whereas Ru metal is obtained at 100°C and above. Ru is being investigated as an electroplating seed layer in electronics, as a capacitor electrode and as a heterogeneous catalyst – all applications that require metal rather than oxide. We show that thermodynamics based on density functional theory (DFT) is a computationally-efficient approach for distinguishing between the possible surface-gas processes. The temperatures and pressures for crossover between different chemistries can be estimated, with the accuracy depending on how entropy, coverage and diffusion are treated. We use DFT to examine the conditions of stability for Ru metal, hydride, hydroxide and oxide with respect to H2 and RuO4 reagents, and so explain the crossover from oxide to metal film just below 100°C. We point out how to balance the cost (in terms of researcher time and computer time) against the benefit that each level of accuracy can offer.
In the second part of the talk, we introduce Microkinetic Modelling, a new Schrödinger capability for examining the overall kinetics of gas-surface chemistry by solving the coupled kinetic rate equations of its constituent elementary reaction steps. This allows the simulation of macroscopic parameters such as sticking coefficients that can be experimentally measured and used as inputs for fluid dynamics simulations. We first outline the computational scheme, where elementary steps and their activation free energies have been computed with DFT. The resulting microkinetic model for alumina ALD yields measurable quantities (e.g. growth rate) as a function of temperature and pressure, which are validated against experiment. Variation with pressure can account for penetration depth and conformality within high aspect ratio features.
The two cases discussed in this talk thus illustrate how atomic-scale DFT can be embedded into higher-level computational schemes for accurate and achievable prediction of the conditions and parameters for controlling chemical processes.
OCT 1 | 18:00 IST
How Physics-based Modeling and Machine Learning Enable Accelerated Development of Battery Materials
Speaker: Garvit Agarwal, Senior Scientist II, Schrödinger
Abstract: The rapid advancements in rechargeable Li-ion battery (LIB) technology over the last decade has revolutionized several key industries such as transportation and consumer electronics. However, new battery chemistries are needed to meet the rapidly growing demand and to improve the power density, safety, reliability, and lifetime of LIBs. Molecular modeling has become an integral part of the design cycle of new battery chemistries. Accurate physics-based modeling enables rapid evaluation and screening of large chemical and material design space thereby, helping industries reduce the time required to bring the new technology to the market.
In this webinar, we will introduce the latest technological innovations in Schrödinger’s digital chemistry platform for battery materials design. In particular, the webinar will focus on examples to demonstrate the application of automated solutions for accurate prediction of thermodynamic stability and voltage profile of cathode materials, ion diffusion pathways and kinetics in electrode materials, transport properties of liquid electrolytes and modeling the nucleation and growth of solid electrolyte interphase (SEI) layers using Schrödinger’s SEI simulator module. We will also introduce an automated generalized framework for the development of customized machine learning force fields for complex materials such as liquid electrolytes, inorganic cathode coatings and solid polymer electrolytes, paving the way for efficient design of novel materials for next generation batteries.
OCT 8 | 18:00 IST
Accelerating the Design of Asymmetric Catalysts with Schrödinger’s Digital Chemistry Platform
Speaker: Saientan Bag, Senior Scientist I, Schrödinger
Abstract: Asymmetric catalysis has become an integral part of the science-driven technological revolution in the second half of the 21st century, leading to decreased energy demands, sustainable chemical processes and the realization of “impossible” transformations. Asymmetric catalysis based on chiral transition-metal complexes plays an important role in the synthesis of single-enantiomer drugs, perfumes and agrochemicals. The importance of the field is recognized by two Nobel Prize Awards in 2001 (transition-metal catalysis) and 2021 (organocatalysis).
Asymmetric catalysts are traditionally designed by experimental trial-and-error methods, which are resource-, time- and labor-consuming, and thus extremely expensive. Digital methods offer the opportunity to expedite catalyst design. Until recently, computational chemistry, typically quantum chemical studies, indirectly contributed to asymmetric catalyst design by providing rationalization for the mechanism of generation of chirality. With the development of more advanced methods, algorithms and an included layer of automation, computational catalysis is now providing the possibility for direct asymmetric catalyst design.
In this webinar, I will demonstrate how Schrödinger’s advanced digital chemistry platform can be used to accelerate the direct design and discovery of asymmetric catalysts.
AI/ML meets physics-based simulations: A new era in complex materials design
Share
The simulation of materials properties using physics-based approaches, such as density functional theory (DFT) and molecular dynamics (MD), has long been successful in providing insights into structure-property relationships and subsequently aiding in the design of novel materials. In recent years, AI/machine learning (ML) has been used extensively in conjunction with physics-based modeling techniques to greatly accelerate materials innovation. The accuracy and generalizability of physics-based modeling improves the performance of AI/ML models and enables them to be used effectively even in small-data regimes. Conversely, the speed and flexibility of AI/ML help bridge the time- and spatial- scale limitations of physics-based models, creating a synergistic approach that optimizes both predictive accuracy and computational efficiency.
In this webinar, we demonstrate the application of this combined approach in designing materials and formulations across diverse materials science applications, from battery electrolytes and fuel mixtures to thermoplastics and OLED devices.
Key Learning Objectives:
Understand how DFT descriptors enhance the accuracy of AI/ML models for optoelectronic molecules and battery electrolytes
Discover how MD simulation descriptors improve AI/ML models for the viscosity of organic molecules
Explore the use of Schrödinger’s automated Formulation Machine Learning solution to:
Train AI/ML models for the solubility of APIs in binary solvents
Predict the motor octane number of hydrocarbons
Learn about advances in AI/ML force field technology (QRNN) and its application in modeling the bulk properties of inorganic cathode coating materials
Our Speaker
Anand Chandrasekaran
Senior Principal Scientist, Schrödinger
Anand Chandrasekaran joined Schrödinger in 2019 and he is currently the Product Manager of MS-Informatics. His expertise is in applying machine learning to different areas in Materials Science and computational modeling. He graduated from the group of Prof.Nicola Marzari in the Swiss Federal Institute of Technology, Lausanne with a PhD in Materials Science. Before joining Schrödinger, Anand also worked in the group of Prof. Rampi Ramprasad on a number of topics including polymer informatics, machine-learning force-fields, and machine-learning for electronic structure calculations.
Schrödinger is excited to be participating in the Festival of Biologics conference taking place on October 15th – 17th in Basel, Switzerland. Join us for a presentation by Esam Abualrous, Principal Scientist I at Schrödinger, titled “Advances in Structure-Based Computational Modeling and Collaborative Enterprise Informatics for Biologics.” Stop by booth 640 to speak with Schrödinger scientists.
Key learning objectives:
Highlight of recent advances in computational structure-based methods for biologics
Address common challenges in antibody engineering.
Illustrate of our collaborative biologics discovery informatics platform
Advances in Structure-Based Computational Modeling and Collaborative Enterprise Informatics for Biologics
Speaker: Esam Abualrous, Principal Scientist I
Abstract: Engineering biologic drug candidates to optimize desirable properties or reduce unwanted characteristics often requires extensive experimentation. This presentation will highlight recent advances in computational structure-based methods rooted in physics and their role in expediting candidate optimization. It will address common challenges in antibody engineering, such as accurately predicting antibody-antigen binding affinities, enhancing structural stability, and identifying and mitigating developability risks. Additionally, the presentation will demonstrate how these workflows are integrated into a collaborative biologics discovery informatics platform. This platform combines experimental data with robust computational modeling, execution, delivery, and analysis capabilities, aiming to expedite and improve decision-making by centralizing all critical information.
On Monday, October 14th at Basel Marriott Hotel, Schrödinger is hosting an immersive workshop on the application of physics-based computational methods (BioLuminate, FEP+) and collaborative enterprise informatics (LiveDesign) towards designing better biologics, faster.
We welcome those who are new to computational tools or those who already have some experience and seek a more structured understanding, including of new and advanced methods. During the session, enjoy a networking lunch to connect with fellow attendees. If you’re interested in attending, please email marketing_europe@schrodinger.com.
Schrödinger is excited to be participating in the BioTechX Europe conference taking place on October 9th – 10th in Basel, Switzerland. Join us for a presentation by Pat Lorton, EVP, CTO, and COO at Schrödinger, titled The Predict-First Paradigm: How Digital Chemistry is Shaping the Future of Drug Discovery. Stop by booth 810 to speak with Schrödinger scientists.
The Predict-First Paradigm: How Digital Chemistry is Shaping the Future of Drug Discovery
Speaker: Pat Lorton, EVP, CTO, COO, Schrödinger
Abstract: Digital chemistry offers a modern paradigm for enabling rapid in silico testing of design ideas using highly accurate computational assays of key properties, accessible across whole project teams. This shift from design strategies based largely on experimental trial and error towards a ‘predict-first’ approach to drug discovery allows teams to dramatically expand the chemical space that can be explored and results in a highly interactive, computationally-driven design-predict-make-test-analyze cycle. Chemists are empowered to test hypotheses through predictive modeling and iteratively improve designs prior to compound synthesis. Teams can confidently explore novel, and often more complex designs while sending only the top scoring molecules for synthesis. In this talk, we will walk through the digital chemistry strategy used by Schrödinger’s therapeutics group, which has led to several successful clinical-stage drug discovery candidates. We will also introduce an upcoming expansion of the computational platform to predict toxicology risk in early stage drug discovery.
Schrödinger is excited to be participating in the HomeCare & Beauty Sustainability Summit 2024 conference taking place on September 11th – 12th in Amsterdam, Netherlands. Join us for a presentation by Jeffrey Sanders, Product Manager and Scientific Lead of Consumer Goods at Schrödinger, titled “Beyond AI: Leveraging physics-based modeling and machine learning to develop sustainable personal care products.” Stop by booth R4 to speak with Schrödinger scientists.
Beyond AI: Leveraging physics-based modeling and machine learning to develop sustainable personal care products
Speaker: Jeffrey Sanders, Product Manager and Scientific Lead of Consumer Goods, Schrödinger
Abstract: The journey to develop and reformulate products to become more sustainable has many challenges. Research and development in these areas often demand substantial time, resources, and new raw materials. To accelerate this process, predictive modeling offers the potential to identify promising ingredients, formulations, and even new packaging materials that meet sustainability requirements. A major obstacle in building and deploying useful models is data sparsity. One promising avenue to explore is multi-scale physics-based simulations, as they do not require large experimental datasets as inputs and can be combined with sparse existing data to generate more robust models. This talk will highlight a case study where physics-based simulation was used to accelerate the development of an eco-friendly personal care formulation, and also how molecular-level simulation can be incorporated into machine learning models when little experimental information is available.
Computationally-Guided Drug Formulation Webinar Series
Date & Time
September 11th, 2024 – May 14th, 2025
Location
Virtual
Share
A smart, strategic drug formulation can efficiently advance your drug development projects and inform downstream processes. Advances in molecular modeling and machine learning are enabling atomistic-level insights and the ability to evaluate large numbers of candidate materials and formulations prior to experiments.
Join us this fall for Computationally-Guided Drug Formulation Webinar Series – seven webinars in which we will explore how the latest computational modeling tools are impacting the various steps in the pharmaceutical formulation process. In each webinar we will feature an expert from Schrödinger sharing valuable insights and practical applications on a key topic. Register for the series to learn how to optimize your formulation process with structure-based insights and efficient parameter screening.
May 14, 2025 Computational insights into polymer excipient selection for amorphous solid dispersions
Speaker: Andrea Browning, Senior Director for Polymers Watch now
April 8, 2025 Accelerating pharmaceutical formulations using machine learning approaches Speaker: Anand Chandrasekaran, Senior Principal Scientist Watch now
November 6, 2024 Modeling lipid nanoparticles: Self-assembly and apparent pKa calculation Speaker: John Shelley, Fellow Watch now
October 23, 2024 Crystal structure prediction workflow for small molecule drug formulation Speaker: Lingle Wang, Sr. Vice President, Scientific Development Watch now
October 9, 2024 Molecular-level insight into solubility-enhancement via cosolvents and amorphous solid dispersions Speaker: Ben Coscia, Principal Scientist Watch now
September 25, 2024 Computational reactivity and catalysis for drug synthesis Speaker: Michael Rauch, Associate Director, Materials Science Watch now
September 11, 2024 Characterizing small drug-like molecules with automated computational spectra prediction Speaker: Art Bochevarov, Research Leader Watch now



 Andrea Browning Director of Polymers and Soft Matter
Andrea Browning Director of Polymers and Soft Matter
















Recent Testimonials