FEB 19, 2026
Introducing RetroSynth: Breaking the synthesis bottleneck with AI and physics-based modeling
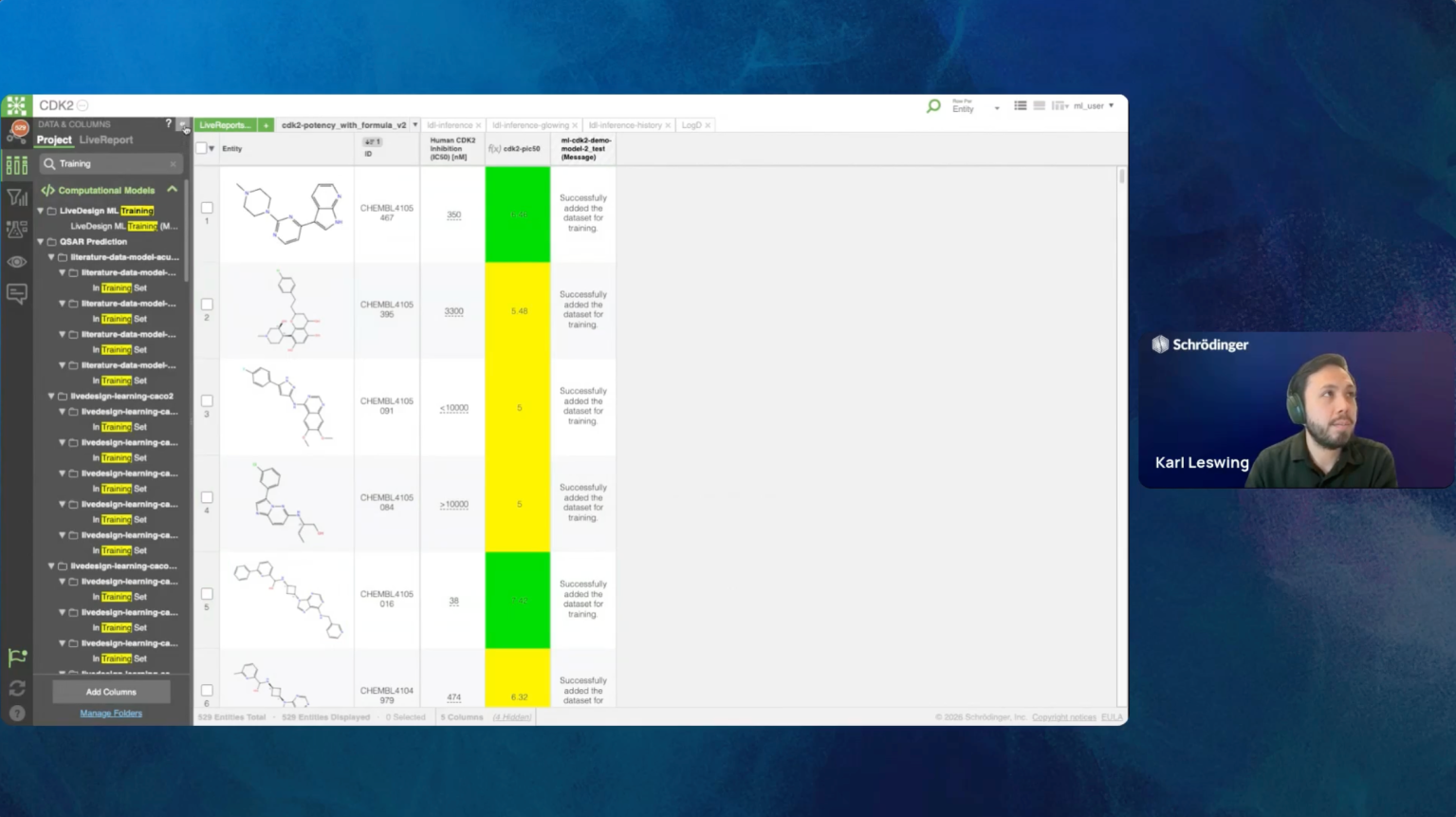
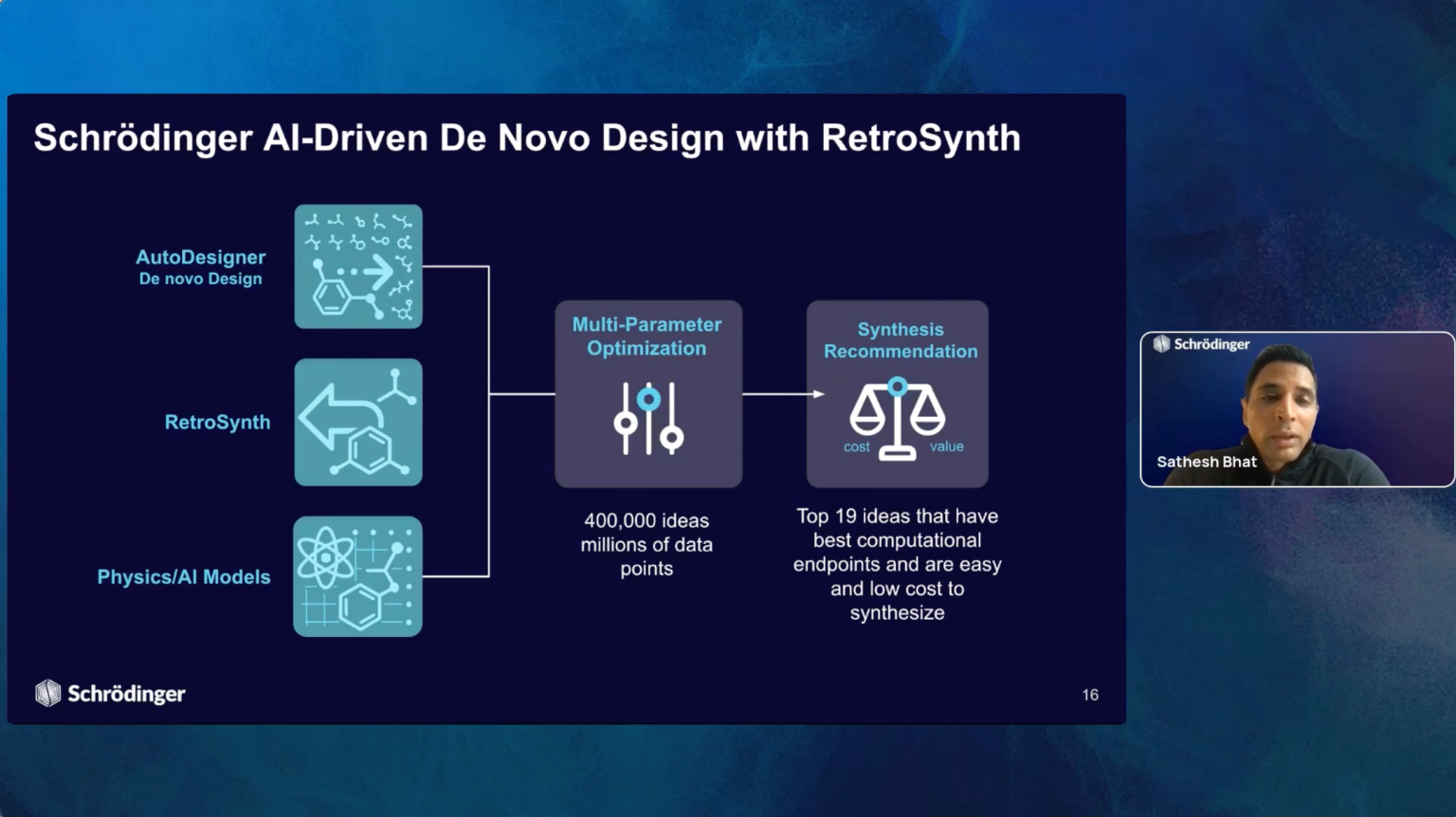
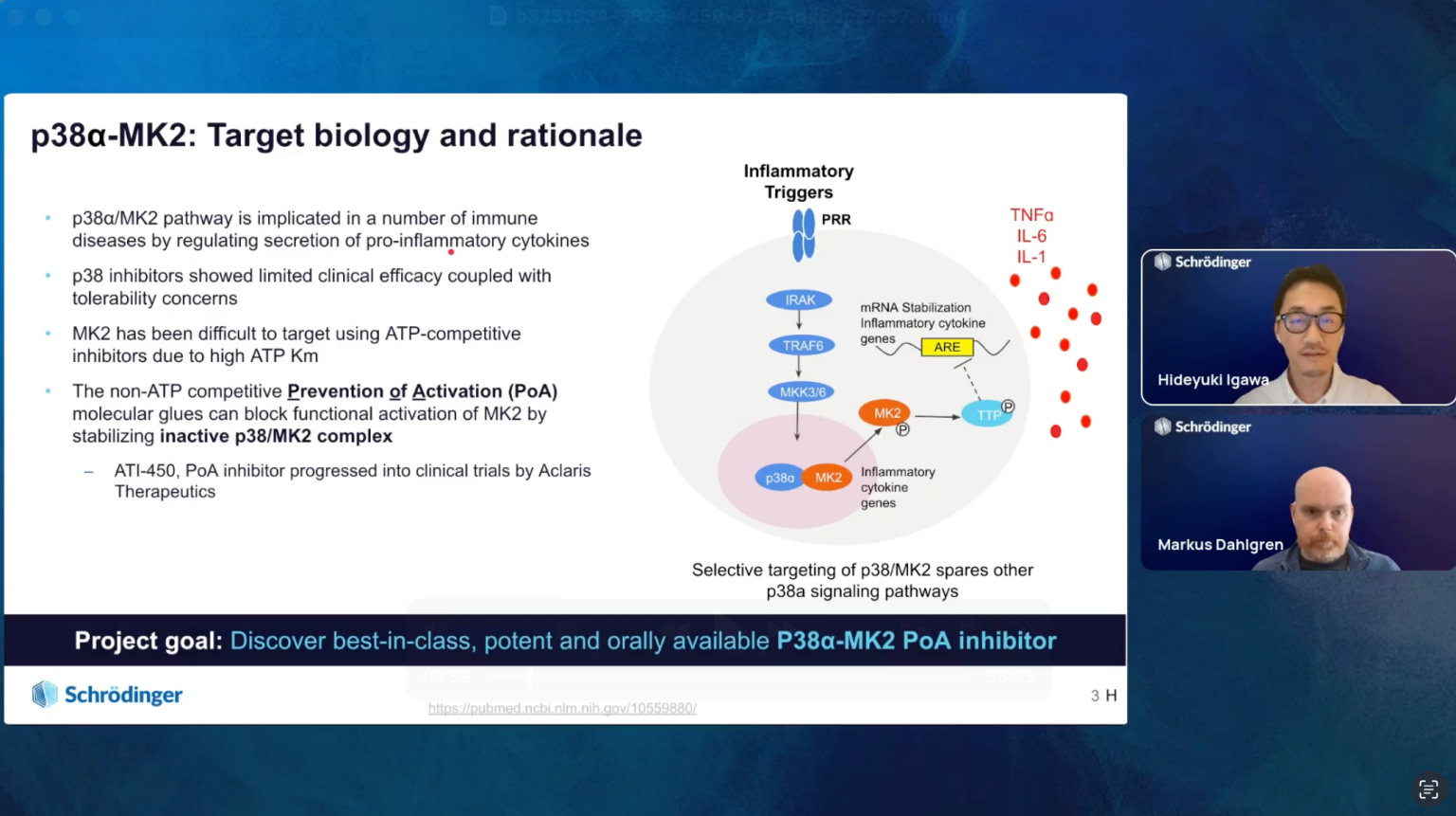
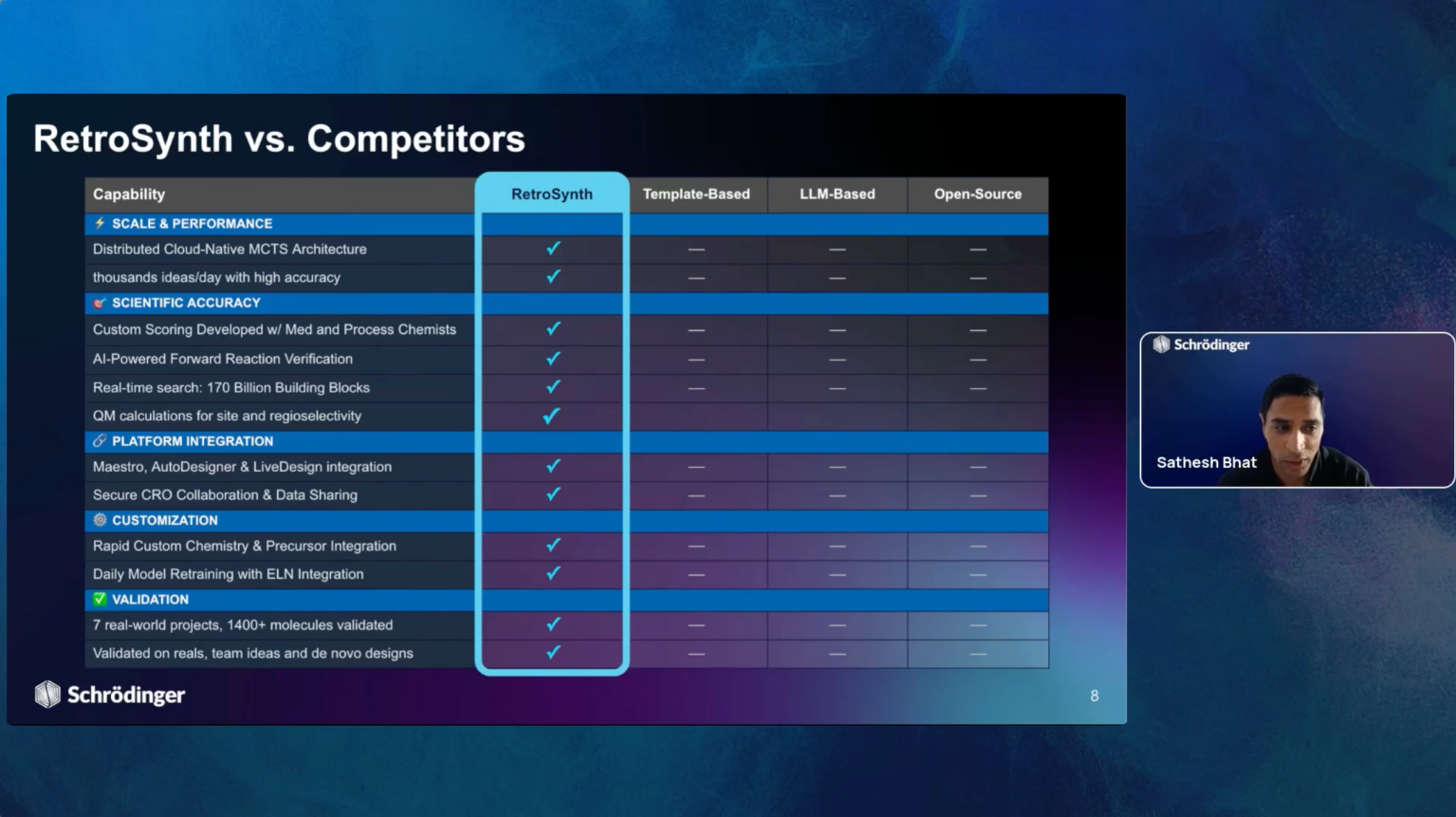
Join us for the introduction of RetroSynth, Schrödinger’s AI-driven synthesis planning platform. RetroSynth is engineered to accelerate and scale conventional retrosynthesis by harnessing advanced deep learning algorithms. RetroSynth uses a distributed cloud-native Monte Carlo tree search (MCTS) architecture to perform highly exhaustive searches to predict and score optimal, accurate, and cost-efficient synthetic pathways. RetroSynth helps you move from complex chemical targets to actionable synthesis plans accurately and efficiently. By integrating real-time building block data with AI and physics-based modeling, RetroSynth unlocks accurate retrosynthetic analysis across millions of de novo designed molecules, leading to massive project acceleration and cost savings in hit identification and lead optimization.
Key Highlights:
- Introduction to RetroSynth: Schrödinger’s product manager will give a detailed description of how RetroSynth works and its advantages over conventional retrosynthesis solutions
- Live Demo: See how RetroSynth performs in action
- Ask questions: Direct technical discussion with our experts
Who Should Attend:
- Medicinal Chemists: Looking to accelerate lead optimization and SAR exploration
- Process Chemists: Focused on identifying scalable and cost-effective synthetic routes early in development
- Computational Chemists: Interested in the integration of ML/AI frameworks into standard R&D workflows
- R&D IT Directors: Evaluating enterprise-grade cheminformatics solutions for chemical synthesis
Our Speakers

Aditya Kaushik
Senior Scientist II, Life Science Software, Schrödinger
Aditya Kaushik is an ML Research Scientist and the lead developer for the generative design and retrosynthesis technologies at Schrödinger. His primary focus is on the research, development, and integration of machine learning approaches to accelerate and optimize Design-Make-Test-Analyze (DMTA) cycles in active drug discovery programs. He received his B.S. from Johns Hopkins University, where he double majored in Computer Science and Chemical & Biomolecular Engineering.

Sathesh Bhat
Executive Director, Therapeutics Group, Schrödinger
Sathesh Bhat, Ph.D., executive director in the therapeutics group, joined Schrödinger in 2011. He is responsible for overseeing computational chemistry efforts on internal and partnered drug discovery programs at Schrödinger. Previously, Sathesh worked at both Merck and Eli Lilly leading computational efforts in several drug discovery programs. He obtained his Ph.D. from McGill University, which involved developing structure-based methods to predict binding free energies. Sathesh has co-authored multiple patents and publications and continues to publish on a wide variety of topics in computational chemistry.

David Papin
Principal Scientist II, Applications Science, Schrödinger
David Papin joined Schrödinger in 2024 as an Application Scientist. David has a background in chemistry and computational chemistry. He held positions in computational sciences in the industry where he provided in silico support for small molecules and large molecules projects.