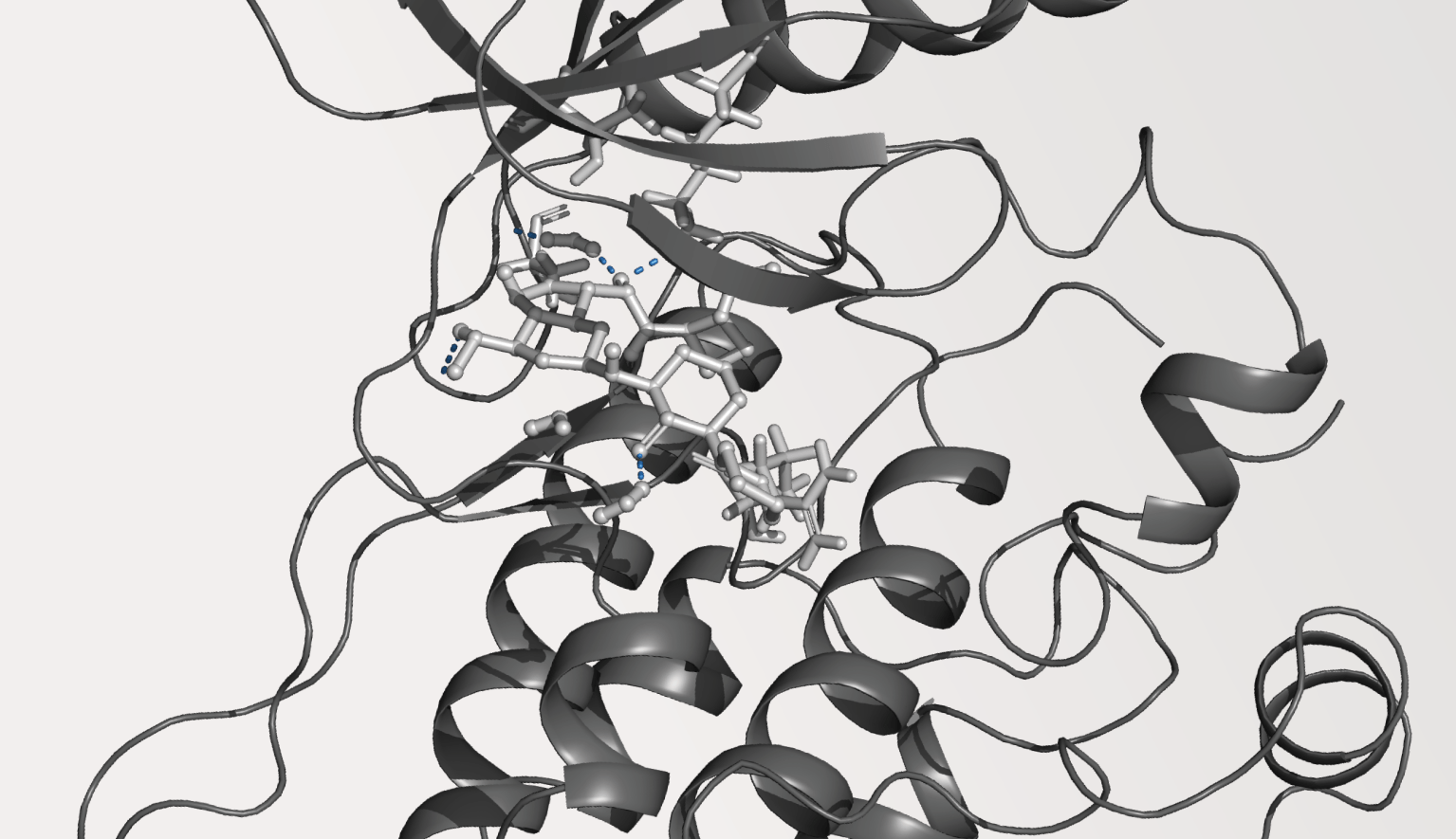SAMPE Europe
- September 24th-26th, 2024
- Belfast, Northern Ireland
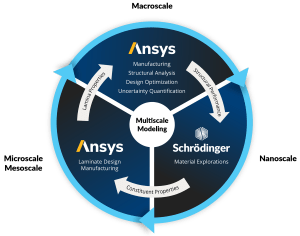
Schrödinger is excited to be participating in the SAMPE Europe conference taking place on September 24th – 26th in Belfast, Northern Ireland. Join us for a presentation by Eli Sedghamiz, Senior Scientist II at Schrödinger, titled “Designing Tailored Sizing Agents for Enhanced Mechanical Properties in Benzoxazine Composites through Molecular Simulation.” Stop by booth 30 to speak with Schrödinger scientists.
Designing Tailored Sizing Agents for Enhanced Mechanical Properties in Benzoxazine Composites through Molecular Simulation
Speaker:
Eli Sedghamiz, Senior Scientist II, Schrödinger
Abstract:
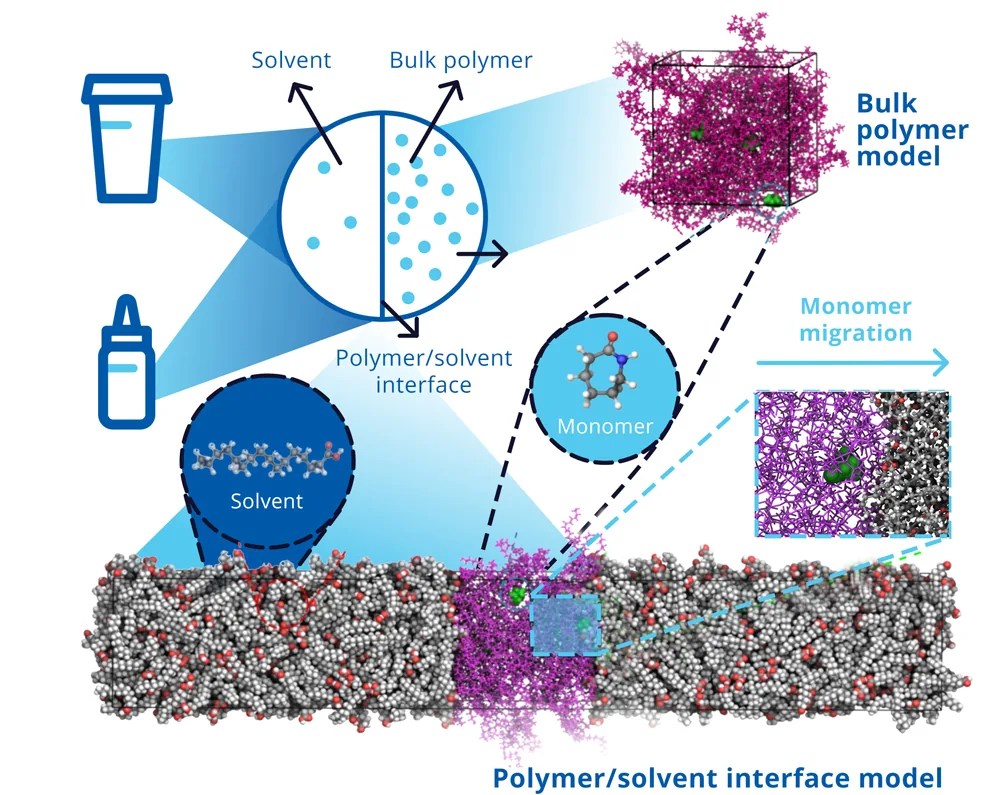
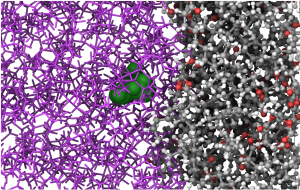
The effective optimization of mechanical properties in composite materials relies heavily on the design of new sizing agents. Our study delves into overcoming challenges encountered in polybenzoxazine as a matrix in carbon fiber composites. While polybenzoxazine boasts high mechanical strength, it often falls short in toughness. Expanding upon experimental insights, we integrate curcumin-based polyurethanes (CPUs) as sizing agents within polybenzoxazine (BA) matrices, while simultaneously adjusting the extent of hydrogen bonding on carbon fiber surfaces. Our aim is to uncover the impact of these adjustments on crucial thermomechanical properties by classical molecular dynamics simulations and to establish structure-property relationships. Our findings highlight enhancements in these properties, emphasizing the transformative potential of tailor-made sizing agents in reshaping composite material performance. This research complements our understanding of composite material design and provides valuable guidance for experimentalists seeking innovative strategies to develop new and high-performance materials.