Pharmaceutical Formulations Workshop 2025
- March 19th, 2025
- 10:00AM CET
- Mannheim, Germany
Using Schrödinger’s Materials Science Suite for molecular modeling and machine learning in the area of pharmaceutical formulation and delivery
Schrödinger invites you to a one-day in-person workshop in Mannheim, Germany to gain hands-on training in the use of our Materials Science Suite for drug development.
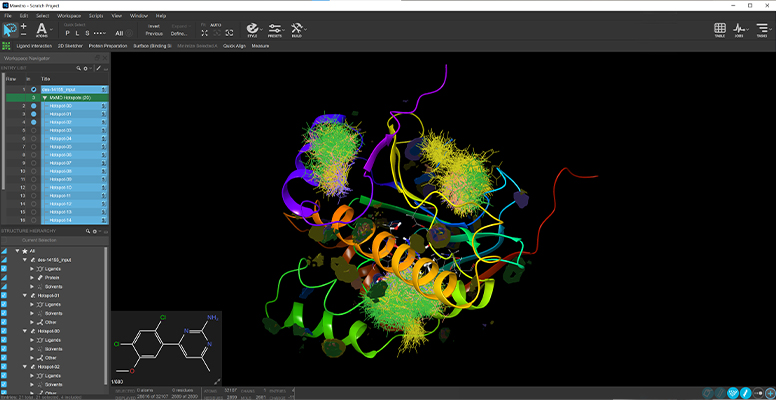
Participants will get practical experience and in-person guidance in using our Materials Science Suite and the tools involved in building molecules, polymers, and complex mixtures for use in molecular dynamics simulations. Leveraging automated property prediction workflows as well as analysis tools will play an important role. Another aspect will be the application of machine learning.
Examples of how molecular-scale simulations can inform drug delivery and formulation research will be included through-out the day.
When & Where:
Wednesday 19th March 2025, 10:00AM CET
Glücksteinallee 25
68163 Mannheim, Germany
(5 minutes walk from Mannheim Hauptbahnhof)
Please see the Agenda for more details about the content presented (Full agenda TBA).
Please see our FAQs for information regarding what to bring, getting to the venue, and accessibility.
If you need further information please contact Patrick Heasman: patrick.heasman@schrodinger.com
If you are interested but unable to attend in person, please reach out to the contact above.
Registration:
Registration is free and includes lunch and refreshments.
Participants must bring their own laptop to access the software, and an external mouse is recommended. We will be utilising our Virtual Computer, which is accessed via web browser – No software installation is required prior to the session.
Places are limited, so please ensure to register as soon as possible.
Registration will close at latest on Friday 14th March 2025
Who should attend:
Any researcher studying drug discovery, design, formulation, or interested generally in learning about computational materials science. No prior experience is required.
Instructional material can be reviewed before or after the workshop for free at:
https://www.schrodinger.com/sites/default/files/s3/release/current/Documentation/html/materials_science/tutorials_TOC.htm
Speakers and demonstrators:
Dr. Caroline Krauter
Dr. Irene Bechis
Dr. Patrick Heasman
Agenda
FAQs
Where is the venue and how can I get there?
The workshop is being help at our offices in Mannheim, Germany. The building is accessible via car, and the train station is within a 5 minute walk.
What is included with my registration?
Registration is completely free to attend the workshop. We will also be providing food and refreshments throughout the day.
Can I join the session virtually / remotely?
As we want to give the attendees help and guidance during the workshop we currently have no intention of running this workshop online. Please reach out if you are interested but are unable to travel to the event location.
What do I need to bring?
A laptop is required for this workshop. We will not be providing any on the day, so please ensure that you bring one. We also recommend that you bring a personal laptop to avoid any firewall restrictions.
An external mouse is not required, but we do recommend that you bring one as our software makes full use of the 3 buttons.
No. We will be utilising the Schrödinger Virtual Computer for all hands-on sessions. A suitable web browser is required for accessing this (Chrome, Edge, Firefox).
How long is the workshop?
The workshop is an all day event to give you the best opportunity to learn about our tools and benefit from the practical sessions throughout. We will start at 10:00 am and finish at approximately 4:00 pm.
For accommodation and travel, we ask that attendees make their own arrangements. There are several hotels within walking distance to the venue, and the train station is situated close by.
Please contact Patrick Heasman (patrick.heasman@schrodinger.com) for any additional information about the event and the location.
Travel:
- Via plane / train: Frankfurt / Frankfurt Airport – A direct train to Mannheim takes approximately 45 minutes.
- Via car: There are several car parks located on Glücksteinallee.
Hotel recommendations:
- Holiday Inn Mannheim City
- LanzCarré Hotel Mannheim
- Premier Inn Mannheim City Centre hotel
- Hilton Garden Inn Mannheim