Release Notes
Release 2025-2
Small Molecule Drug Discovery
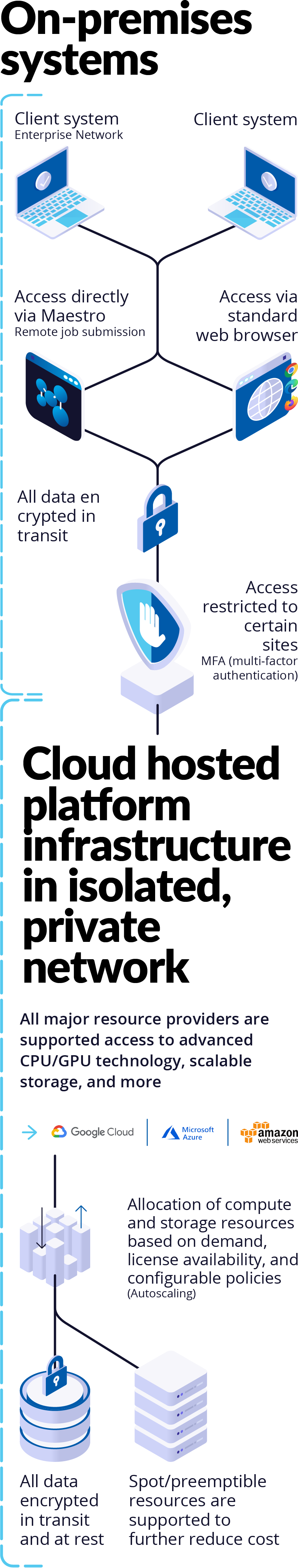
Platform Environment
Maestro Graphical Interface
- New Welcome Screen on startup provides quick access to common tasks such as creating and opening projects and importing structures
- Modernized and streamlined Project Table for enhanced usability
- New Table Configuration pane allows fast switching between Light and Dark themes and toggles visibility of the ePlayer and Property Tree.
- New Gadgets Menu provides convenient access to Charts and the 2D Viewer
- New Workflow Action Menu (WAM) to view spectroscopy results from Jaguar and Jaguar Spectroscopy calculations in the Project Table
Target Validation & Structure Enablement
Protein Preparation
- Improved minimization protocol to support broader coverage of biological and chemical systems
- Produce more reliable prepared structures by expanded coverage of equivalent tautomeric ligand states
- More easily view serious structural issues by filtering diagnostic reports with a severity threshold
- New ‘Missing Atom’ tab on the Diagnostics panel enables select sidechain and loop modeling
Cryo-EM Model Refinement
- GlideEM poses are now sorted by GlideScore which is more discriminating in ranking low RMSD structures than Denscore
Ligand Preparation
Ligand Docking
- Faster Glide scoring and docking with optimized Glide (Beta): Screen larger libraries and find better candidates with optimized Glide, including enhanced Active Learning Glide and Python API support
- Same industry-leading Glide docking funnel and scoring functions, Emodel and GlideScore
- Faster turnaround with same compute resources for Active Learning Glide and AutoDesigner
- Advanced Python API support offers easy automation and file control over docking process for greater experimentation
- Accessible through the new Ligand Docking panel that enables setup of Active Learning and Glide calculations
ABFEP
- Energy Decomposition data is now reported in Analysis PDF reports
Lead Optimization
FEP+
- New FEP+ Pose Builder workflow for automatically generating high-quality ligand alignments (Beta): Generate FEP-ready poses faster and run FEP+ at scale with an automated workflow designed for unbiased selection and robust atom-mapping
- Ability to read and write FEP+ Protocol files directly in the FEP+ Panel
- Improved Classification matrix styling
- Kendall’s tau statistic added to the statistical metrics reported
- Improvements to exported FEP+ data in csv/xls formats
- Added ‘None’ as a new Hot Atom Rule
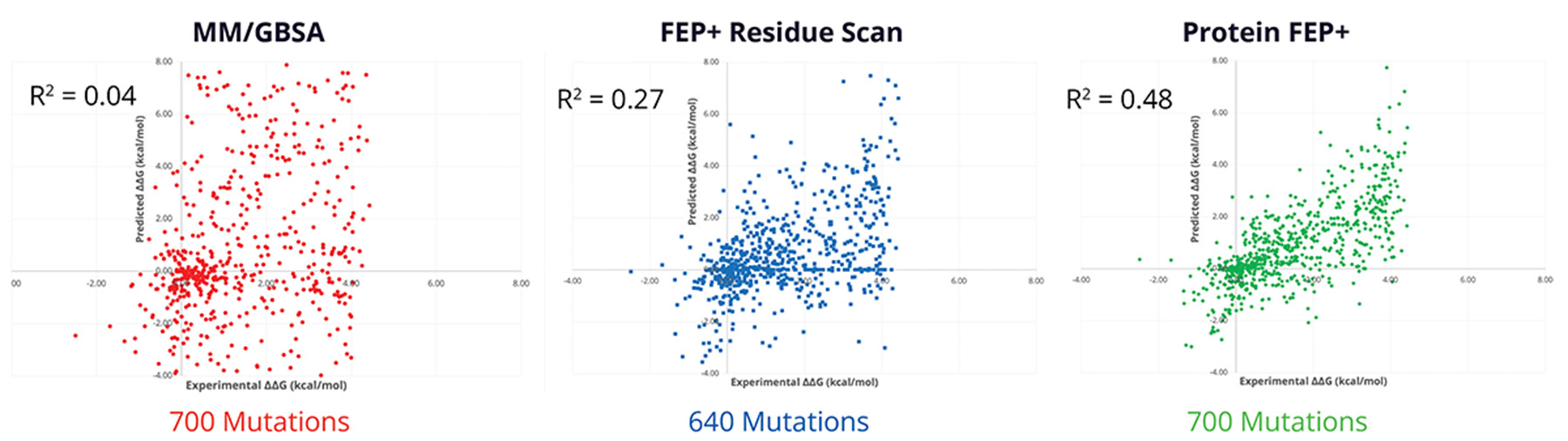
Protein FEP
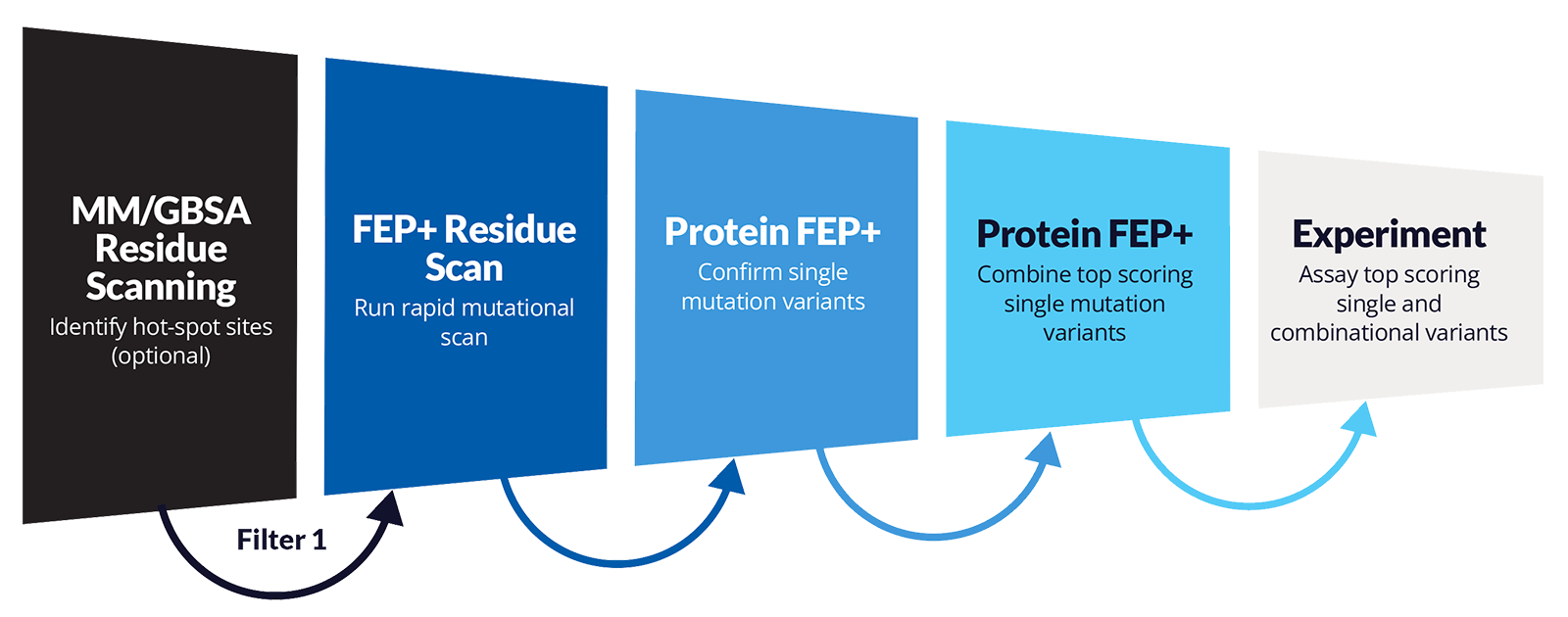
- FEP+ Residue Scan supported in Protein FEP+ for Ligand Selectivity panel
Constant pH Simulations
- Added support for Cysteine residues
FEP+ Protocol Builder
- Sharply reduced compute resources to run default workflow by shrinking initial simulation times to 0.5 ns and extended times to 10 ns
- Seamless interconnection as FEP+ Panel can now Read/Write Protocol Builder files
- Bias the selection of protocols to extend including compute efficiency via Pareto analysis (command line only)
- Added support for covalently bound ligands
- Ability to optionally sample charge states of GLU, ASP, LYS, ARG, and CYS in protocol optimization
De Novo Design
AutoDesigner – R-group Design
- New R-group Similarity score feature to focus ideation around compounds of interest
- New Design Rationale capability to improve ADME endpoints with respect to reference ligands
Alternative Modalities
Bifunctional Degraders
- Expanded support for protein degrader modeling with the new Degrader Sampling Workflow (Beta): Generate accurate degrader ternary complexes through integration of protein-protein docking and linker sampling in a structure-based workflow
Biologics Drug Discovery
- Augmented AI/ML capabilities for biologics with machine learning-based T-Cell Receptor (TCR) structure prediction (Beta): Perform high throughput structure prediction and large scale modeling of TCRs with the ImmuneBuilder deep learning model and Prime
- New Macromolecular Pose Filtering panel to filter native or near native poses from an ensemble of complexes using experimental data such as HDX-MS
Materials Science
GUI for Quantum ESPRESSO
Product: Quantum ESPRESSO (QE) Interface
- A new environment variable for the location of Quantum ESPRESSO binary
Transport Calculations via MD simulations
Product: MS Transport
- Thin Plane Shear: Selection of slab region by molecular units
KMC Charge Mobility
Product: MS Mobility
- Compute KMC Charge Mobility: Predictions based on Schrödinger’s new mobility engine
Materials Informatics
Product: MS Informatics
- Machine Learning Property: Updates to existing models
- Machine Learning Property: Prediction of triplet reorganization energy
- Machine Learning Property: Prediction of S1-to-T1 energy gap (∆EST)
- Machine Learning Property: Predictions from the interactive mode automatically added to the Project Table
- MLFF Calculations (Beta): Single-point energy and geometry optimization tool using Schrödinger’s latest machine-learned force fields
Formulation ML
Product: MS Formulation ML
- Formulation ML: Support for custom ingredient descriptors
- Formulation ML: Support for creating models using multiple CPUs in parallel
- Formulation ML: Support for setting mixtures as individual components
- Formulation ML Optimization: Workflow solution to optimize materials formulations
Layered Device ML
Product: MS Layered Device ML
- OLED Device ML: Workflow solution to predict OLED device performance
- Optoelectronic Device Designer: Use ML OLED device models to predict performance
Coarse-Grained (CG) Molecular Dynamics
Product: MS CG
- Automated CG Mapping: (+AUTOMAPPING_MARTINI_PROTEIN) Support for proteins in automated mapping and parameterization for Martini
- Automated CG Mapping: Accurate mapping for carbohydrate systems
- Improved threshold for momentum errors in CGMD simulations
- CG FF Builder: Parameters for water-water interactions fixed by default
Dielectric properties
Product: MS Dielectric
- Complex Permittivity: Option to run replicates in parallel
Reactivity
Product: MS Reactivity
- Reaction Network category created under the Materials task menu
- Reaction Workflow renamed to Reaction Network Profiler
- Auto Reaction Workflow renamed to Reaction Network Enumeration Profiler
- Reaction Network Profiler: Option to run conformational search using CREST
- Reaction Network Profiler: Conformational search included in restarts (command line)
- Nanoreactor: Option to screen products by energy relative to reactant state
- Nanoreactor: (+ELEMENTARY_REACTION_NETWORK) Support for the Elementary Reaction Network workflow
Microkinetics
Product: MS Microkinetics
- Microkinetics Deposition Analysis: Workflow solution to run post-analysis of microkinetic simulations in deposition or etch processes of solid materials
- Microkinetic Modeling: (+MATSCI_MKM_INTERACTIONS) Support for simple quadratic adsorbate-adsorbate interactions
Reactive Interface Simulator
Product: MS RIS
- Solid Electrolyte Interphase: Option to block intramolecular reactions (command line)
- Solid Electrolyte Interphase: Option to use DFT charges for new species
Crystal Structure Prediction
Product: Crystal Structure Prediction
- Crystal Structure Prediction: Interface and workflow to predict crystal structures and polymorphs for a given molecular compound
MS Surface
Product: MS SurfChem
- Adsorption Enumeration: Access to workflow assessing reactive adsorption
- Desorption Enumeration: Workflow solution for assessing desorption of multiple molecules
MS Maestro User Interface
- Direct link from the task menus to Materials Science Panel Explorer page
MS Maestro Builders and Tools
- Structured Liquid: Automatic standardization of custom lipids
- Polymer: Improved dihedral setups for multiple shortest-length backbones
- Organometallic Conformational Search: Option to run conformational search using CREST
Classical Mechanics
- Evaporation: Option to export the results as CSV file
- MD Multistage: Center of mass motion removed for coarse-grained systems
- Thermophysical Properties: Option to save trajectory energy file
- Umbrella Sampling (Beta): Workflow solution for umbrella sampling of membranes
Quantum Mechanics
- Adsorption Energy: Support for reactive adsorption and desorption energies
- Adsorption Energy: Improved assessment of entropy loss during the adsorption
- Bond and Ligand Dissociation: Option to set product charges from formal atomic charges
- Bond and Ligand Dissociation: Support for PCM and SMD solvent models
- Bond and Ligand Dissociation: Improved 2D visualization of charges and radicals in product fragments
- Crest: UI for semiempirical QM based conformational search using CREST
- Optoelectronic Film Properties: Support for multiple reorganization energies as input for computing intersystem crossing (ISC) rate
- Optoelectronic Film Properties Viewer: Support for user-input reorganization energies to instantly re-evaluate SEET rate
- Thermochemistry Viewer: Support for viewing reactive adsorption and desorption energies
- Trajectory Density Analysis: Improved naming scheme for atom groups
Education Content
Life Science
- New tutorial: Exploring Protein Binding Sites with Mixed-Solvent Molecular Dynamics
- New tutorial: Introduction to T-Cell Receptor Modeling with BioLuminate
- Updated tutorial: Antibody Visualization and Modeling in BioLuminate
- Updated tutorial: Peptide Modeling with BioLuminate
- Updated tutorial: Target Analysis with SiteMap and WaterMap
- New QRS: Structure Reliability Report
- New QRS: Custom Reactions for Covalent Docking
- New QRS: Mixed-Solvent Molecular Dynamics
- Updated QRS: GlideWS Model Generation
- Updated QRS: MM-GBSA Residue Scanning
Materials Science
- New Tutorial: Umbrella Sampling
- New Tutorial: Crystal Structure Prediction
- New Tutorial: Optimization of Formulations Using Machine Learning
- New Tutorial: Machine Learning for OLED Device Design
- New Tutorial: Nanoemulsions with Automated DPD Parameterization
- New Tutorial: Applied Machine Learning for Formulations
- Updated Tutorial: Atomic Layer Deposition
- Updated Tutorial: Design of Asymmetric Catalysts with Reaction Network Enumeration Profiler (previously AutoRXNWF)
- Updated Tutorial: Machine Learning Property Prediction
- New QRS: CREST
- New QRS: Microkinetics Deposition Analysis
LiveDesign
What’s Upcoming in 2025-2
- Enhanced support for antibody-drug conjugates: Import ADCs from a source database and view the entire subcomponent hierarchy in the spreadsheet
- Improved collaboration with freeform column comments: View a comment thread in the main spreadsheet and enable a structured, context-specific conversation with user attribution and timestamps
- Ability to standardize workflows with form templates: Create a standardized data view or analysis, and add the Form to other LiveReports
- New and updated protocols: The FEP+ Pose Builder Protocol has been updated and a new FEP+ Amenability Protocol returns additional pose quality metrics.
Training & Resources
Online Certification Courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Tutorials
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.