
Release Notes
Release 2024-4
Small Molecule Drug Discovery
Platform Environment
Maestro Graphical Interface
- Improved usability in scatter plots and histograms:
- See relationships in data across multiple plots and histograms with streamlined menu into “Entry Actions” and “Sync Options” menu icon
- Added support for string and boolean histograms
- In the histogram panel, easily switch between settings and data table views
- Specify the number of columns and rows for fine control of Workspace Tiles
- Save animated GIF of vibrational motion from Jaguar frequency calculation
- Enhanced Cryo-EM surface performance:
- Up to 2x faster loading of Cryo-EM surface files
- Up to 5x increase in speed for isosurface contour creation and adjustments
- Refined toolbar design for enhanced simplicity, modern aesthetics, and optimization for dark mode
- Updated Maestro Project format to version 5:
- Support for multi-letter chain names beyond traditional 26 characters
- Compressed .prjzip files designed for easy sharing via email
- Automatic conversion of version 4 projects to version 5 upon opening
- By default save Maestro Projects in version 5 format with an option to save in version 4 for backward compatibility
- Support added for two new CIF file formats: “PDBx/mmCIF (*.cif)” and “Small Molecule CIF (.cif)”
- Opening Maestro locally from LiveDesign is now supported on macOS, Linux, and Windows
- Revamped splash screens & iconography: Modern visuals for an updated look and feel
Workflows & Pipelining [KNIME Extensions]
- LiveDesign Admin node can take user credentials from the LiveDesign Connection node enabling SSO configuration
Target Validation & Structure Enablement
Protein Preparation
- Protein The Protein Preparation Workflow now considers Epik states of ligands during the hydrogen-bond network optimization stage by default
Protein X-Ray Refinement
- New sf2map.py script quickly generates an aligned x-ray map, given an input structure and a cif file containing structure factors
IFD-MD
- Updated IFD-MD for automatic sampling of histidine tautomer states (HID, HIE): Consider induced fit effects simultaneously to resolve receptor tautomeric states and predict receptor and ligand conformations
Binding Site & Structure Analysis
SiteMap
- Automatically apply Combined Mode which breaks down sites larger than 800 Å3
Mixed Solvent MD (MxMD)
- Improved cryptic pocket identification with new mixed solvent molecular dynamics (MxMD) interface including customizable visualization (Beta): Gain a clearer understanding of candidate binding pockets on the protein surface with a new interface to set up and analyze MxMD simulations
Hit Discovery
Active Learning Applications
- Faster time-to-results in AL-Glide and Glide using ZeroMQ mode for machine learning evaluation stage and Glide docking stage
Shape Screening
- Additional similarity normalization schemes now available for Quick Shape and 1D Screening command. In addition to the max{O(A,A), O(B,B)} default can apply min{O(A,A), O(B,B)}, O(A,A), and O(B,B) where O(A,B) is the overlap between ligands A and B
- Run Quick Shape and 1D Screens against a Phase pharmacophore hypothesis as the query
- Improve speed of Quick Shape calculations with -limit and -NJOBS1D options that enable more efficient utilization of compute resources
Glide
- Full release of Glide WS mode, previously known as WScore, to prioritize ligands for improved hit enrichment and pose prediction accuracy
- Leverages explicit water energetics to enhance the accuracy of protein-ligand poses and reduce experimentally inactive compounds in top-scoring virtual hits
Lead Optimization
FEP+
- Gain deeper insights into receptor-ligand interactions with new Per-Residue Energy Decomposition in FEP Edge analysis
- Perform categorical analysis in the Correlation Plot (FEP+) interface using classification matrices including common metrics such as Accuracy, Specificity, Recall, Precision, F1 Score, Cohen’s K and Kendall’s T
- View reason why compounds are skipped in ABFEP calculations in the FEP+ Panel
Protein FEP+
- Full release of Protein FEP+ Residue Scanning (with lambda dynamics)
- Workflow now generates an FMP archive to be loaded in the FEP+ Panel
- Web Services support
FEP+ Protocol Builder
- Ability to run with either OPLS4 or OPLS5 force field
- Added support for sampling of more residue protonation states
- New option to skip active learning and perform exhaustive exploration of protocol parameter space
Quantum Mechanics
- Return solvation entropy in implicit solvent calculations
distributed_frequencies.py workflow for numerical frequency calculations - Added support for isotope 11B in NMR calculations
- Implicit solvent model SMD now has gradients and frequencies
- E-sol now supports the CPCM-X implicit solvation method for rapid solvation energies (command line only)
Semi-Empirical Quantum Mechanics
- Updated xTB to version 6.7.1 which uses the advanced solvent model CPCM-X
Macrocycles
- Expanded list of predefined linkers from 5 to 22 for small molecule cyclization in macrocycle.py
- Expanded list of side-chain bridges for peptide cyclization from 4 to 21 of the most commonly reported in the literature
- Control spacers in macrocyclize.py via a CSV file of SMILES strings
- Improvements to macrocycle alignment reproducibility and performance in tug_align script and Ligand Alignment Panel
- Macrocycle sampling script can optionally output only macrocycle conformers
- Easily create cyclic peptides from sequence on the command line with peptide_cyclize.py script
De Novo Design
AutoDesigner – R-group Design
- Boost exploration of similar ligands with new AutoDesigner Similarity feature that scores output ideas based on similarity to a user-provided set of compounds
- Added exhaustive PathFinder enumeration of all routes of the starting ligand using all available building blocks for those routes
- Added recursive trimming of the final set of outputs to generate additional outputs
- Improved logging including an overview of the number of compounds generated at various stages of the workflow
AutoDesigner – Core Design
- Improved logging including an overview of the number of compounds generated at various stages of the workflow
Biologics Drug Discovery
- Predict protein properties with automated machine learning model building using protein descriptors: Leverage AutoQSAR analysis to train, validate, and apply AI/ML models for biologics properties prediction
- Predefined selection sets for quick access to TCR regions like CDRs, alpha/beta chains, and more
Materials Science
GUI for Quantum ESPRESSO
Product: Quantum ESPRESSO (QE) Interface
- Phonon-dependent dielectric properties reported in the Phonon DOS viewer
- Workflow action menu (WAM) for NMR calculations
- Support for phonon calculations with DFT-D3
- Improved cell relaxation protocol
- Schrödinger-compatible Quantum ESPRESSO releases available at Github
- Support for distributed phonon calculations
- Control over maximum number of retries after failure via config file (command line)
- Initial parameters and constraints preserved in the QM Convergence Monitor
KMC Charge Mobility
Product: MS Mobility
- Compute KMC Charge Mobility: Improved speed with robust QM convergence (command line)
Materials Informatics
Product: MS Informatics
- Formulation ML: Increased number of available steps for hyperparameter tuning
- Formulation ML: Option to replace hyperparameter tuning steps with training time
- Formulation ML: Visualization of atomic contributions from the feature importance analysis
- Machine Learning Property: Updates to existing models
- Machine Learning Property: Prediction of melting point for molecular solids
- Machine Learning Property: Prediction of non-aqueous solubility of molecules
Coarse-Grained (CG) Molecular Dynamics
Product: MS CG
- Automated CG Mapping: Speed up for mapping large molecules
- Automated CG Mapping: Particle types and the number of occurrences reported
Penetrant loading simulations
Product: Penetrant Loading (PL)
- Penetrant Loading: Differentiation between pre-existing water and added water
Reactivity
Product: MS Reactivity
- Nanoreactor: Option to set the width of the biasing potential
- Reaction Workflow: Anharmonic zero point energy (ZPE) added to Project Table
- Reaction Workflow: Support for enumeration on sites in rings
- Reaction Workflow: Option to automate the swap fragment with enumeration
- Reaction Workflow: Preview of reaction diagram at the setup
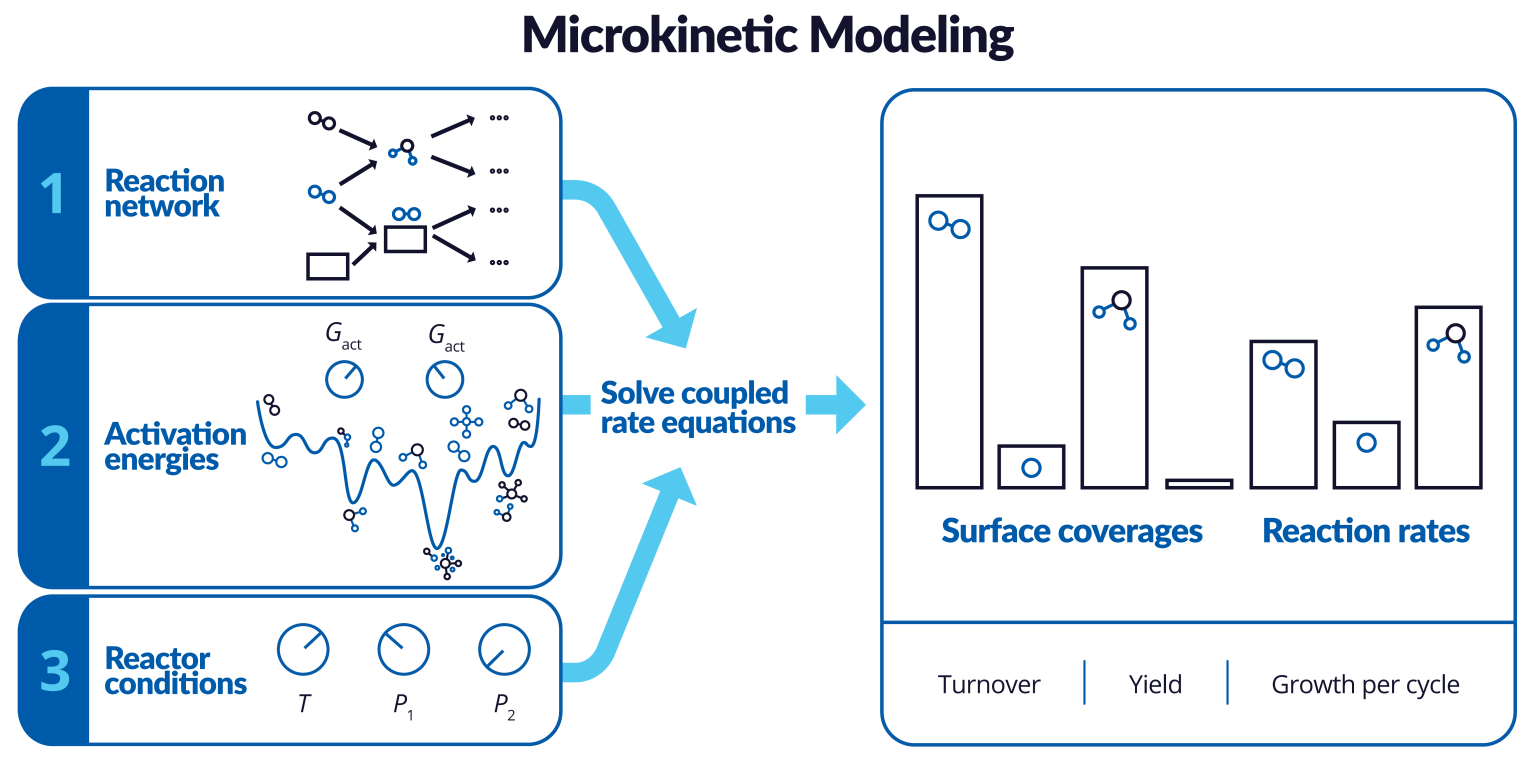
Microkinetics
Product: MS Microkinetics
- Microkinetic Modeling: Support for multistage MKM analysis
- Microkinetic Modeling: Support for zoom in on plots in the viewer panel
- Microkinetic Modeling: Increased default value for maximum integration time step
MS Maestro Builders and Tools
- Adsorption Enumeration: Support for selection of reactive atoms by atom indices
- Disordered System: Improved UI with reconfigured options for tabs and dialogs
- Disordered System: Support for keeping selected molecules rigid with tangled-chain option
- Meta Workflows: Support for radial distribution function analysis
- Nanoparticle: Option to include only molecules with center of mass inside the particle
Classical Mechanics
- Barrier Potential for MD: Option to remove barrier from input structures
- Droplet: Support for entering random seed in building a droplet
- Droplet: Support for randomized initial velocities
- Evaporation: Support for full control over which profiles to plot
- Evaporation: Support for applying barrier potentials
- Evaporation: Option to set evaporation zone based on the distance from COM of the substrate
- Evaporation: Improved loading speed for large input structures
- MD Multistage: Improved relaxation protocol for ladder polymers
- Refined default timestep for DPD particles
- Thermostat and barostat settings set automatically for atomistic and coarse-grained systems
- Stress Strain: Option to plot normal average stress
- Thermophysical Properties: Option to return *.ene files (command line)
- Trajectory Density Analysis: Output *.csv files
Quantum Mechanics
- Adsorption Energy: Option to select between kcal/mol and kJ/mol for energy units
- Adsorption Energy: All output entries incorporated in Project Table as subgroups
- Adsorption Energy: Robust detection algorithm for valid input adsorbates
- Adsorption Energy: Support for loading options from a Quantum ESPRESSO config file
- Optoelectronic Film Properties: Prediction of molecular refractive indices
- Optoelectronic Film Properties: Prediction of intersystem and reverse intersystem crossing rates
- Optoelectronic Film Properties: Improved loading protocols for large input structures
Education Content
Life Science
- New Tutorial: Protein pKa Prediction with Constant pH Molecular Dynamics
- Updated Tutorial: Glide WS Evaluation of HSP90 Ligands
Materials Science
- New Tutorial: Singlet-Triplet Intersystem Crossing Rate
- New Tutorial: Modeling the Formation and Decomposition of Nitrosamines
- New Tutorial: Atomic Layer Deposition
- New Tutorial: Elemental Enumeration
- New Quick Reference Sheet: Refractive Index
- Updated Tutorial: Introduction to Multistage Quantum Mechanical Workflows
Docs Content
- New documentation page to explore solutions for materials science applications and to identify the best fit for users’ interest
- Panel images shown in the help topic of each panel
LiveDesign
What’s new in 2024-4
- Biologics SAR Visualization: View, highlight, and analyze properties alongside sequence differences in the Sequence Viewer tool
- Ligand Designer
- Upload multiple overlays in the Ligand Designer Configurations via the admin panel, and enable or disable the overlays during design sessions
- Rename ligands after editing and using the “predict pose” functionality to track and manage iterative design changes
- R-group enumeration: Filter output products by computed properties
- LiveDesign Learning: View the LiveDesign Learning Dashboard within Landing Pages
-
*LiveDesign Learning is now called LiveDesign ML
-
- LiveReport Management
- View row, column, and cell count for LiveReports in the LiveReport Picker
- Set up a LiveReport as Read-Only or Hidden during creation in an updated Create LiveReport dialog
- The following menu items have been consolidated into an “Edit LiveReport…” menu: Rename, Move to Folder, Make Read Only and Make Editable, Make Hidden and Make Visible
- Admins can update Read-Only LiveReports to make them editable
- Unhide a subset of compounds by clicking on a link in the LiveReport footer and selecting the compound IDs in a dialog
- Documentation
- The ? button in LiveDesign now directs to online documentation as is done elsewhere in the Schrödinger platform. Users will be directed to log into their Schrödinger web account that will be verified with a code to their email; if they do not have a web account, they will need to sign up for one.
- Copy values from the Form spreadsheet, table, and ID widgets to the clipboard
- Search and filter for a column name in the Create MPO dialog when adding a constituent column from the LiveReport
- Click a link in a 3D cell for a 3D Generic Entity and Explicit Ligand Designer to view the 3D structure in Maestro
- Entities appear in a LiveReport more quickly after a reaction enumeration, R-group enumeration, file upload, Maestro upload, and LDClient upload
- LDClient’s API for retrieving FFC columns “get_freefrom_column_by_id” will now include an “updated_at” field containing a long corresponding to the timestamp that the FFC was last updated
- Landing Page: View recent published experimental data from the Compound’s Page
What’s Been Fixed
- Adding an entity to an Advanced Search, by searching for its ID, would show an unresponsive dialog, and now shows a dialog that accept an ID.
- Complex filters would not accept a pasted biologic sequence, and would fail to filter to that sequence, but now accept pasted biologic sequences.
- Creating a LiveReport from a template in the Landing Page would not open the newly created LiveReport, and now opens the LiveReport in a new browser tab.
- If an entity is imported to multiple projects, purging the entity from one of the projects will make it disappear from that project only. Only one file entry now appears in the Manage Files Dialog for a multi-entity import through CSV, ZIP, and FASTA.
- String type of entity relationship metadata is supported in DI by adding a DI mapping for relationship and relationship metadata.
- The column “Structure Class” has been renamed to “Entity Type” in the Data & Columns tree
- Pasting aromatic structures into the sketcher would flip chirality on structures containing a pyrrole, and now the chirality is maintained.
- When entering FFC date values via LDClient, users must now use the YYYY-MM-DD format. If the date is entered in an incorrect format, the system will return a 400 error with the message: ‘Date must be in YYYY-MM-DD format.’
- Forms kanban widgets would show a pin icon beside the kanban tile, and now pin icon does not appear in kanban widgets.
- Unpinning a row now works as expected.
- Model results with multiple values in a cell would show a different order of values if the LiveReport was duplicated, and now show the same order in the duplicated LiveReport as the origin LiveReport.
- The Guanidine group in Arginine incorrectly displayed a carbon with five bonds in the 3D visualizer, and now accurately represents the chemistry, showing the correct bonding structure for Arginine residues.
- Reordering of R-group scaffolds will work for newly added or deleted scaffold without page refresh in SAR analysis(R-group decomposition).
- The deleted docked poses will no longer reappear, ensuring a clean and organized workspace. Each new pose generated after clicking “predict pose” will be sequentially named, allowing for easy tracking (e.g., “docked_ligand_4” following the deletion of “docked_ligand_3”).
- Changing the model’s name via Admin panel will get correctly reflected in the Ligand Designer.
- Targets would remain visible in the 3D visualizer after unchecking the display checkbox and then selecting a different entity, and now the Target display checkbox remains unchecked and the Target is not visible.
- Copying a LiveReport from one project to another would grant the destination project ACLs to any model within the LiveReport, even if the Model’s Protocol did not provide access to the destination project. Now, the model column will be “dummified” and not visible in the destination project if the Protocol does not provide access to that project.
- Models with dependent parameterized models will not show an error when archived, and all of its dependent parameterized models will also get archived. A dialog box will appear stating the number of dependent parameterized models that will be archived.
Training & Resources
Online Certification Courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
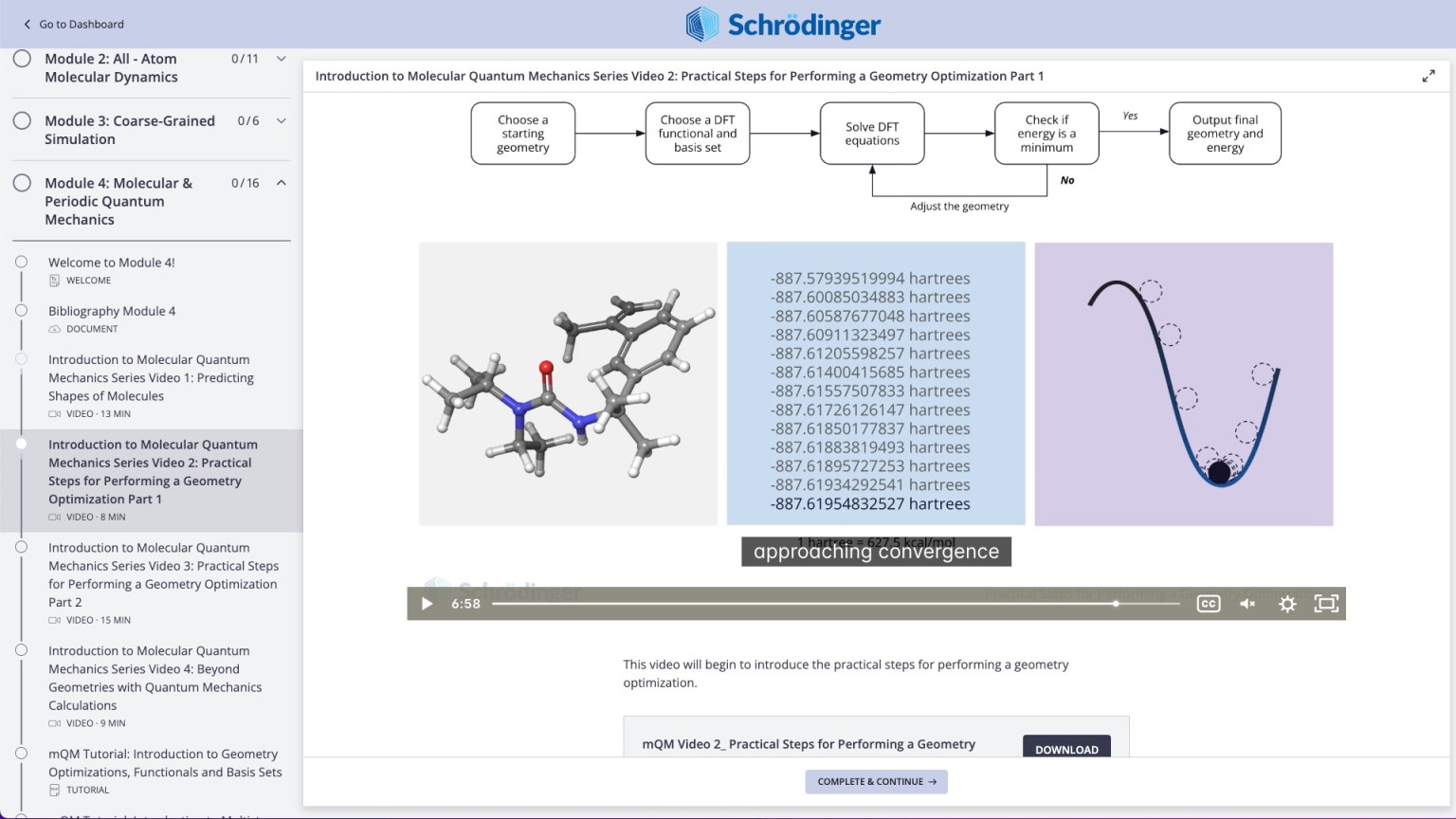
Tutorials
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.


































Recent Testimonials