OLEDs: Innovations, Manufacturing, Markets
- April 10th-11th, 2024
- 8:00am PDT
- Virtual
Schrödinger is excited to be participating in OLEDs: Innovations, Manufacturing, Markets taking place on April 10th – 11th online. Join us for a presentation by Hadi Abroshan, Principal Scientist and Product Manager at Schrödinger, titled “Revolutionizing Organic Electronics: Computational Insights and Innovations in OLED Materials Design.”
Speaker:
Hadi Abroshan, Principal Scientist and Product Manager
Date/Time:
April 11 | 8:00am PDT
Abstract:
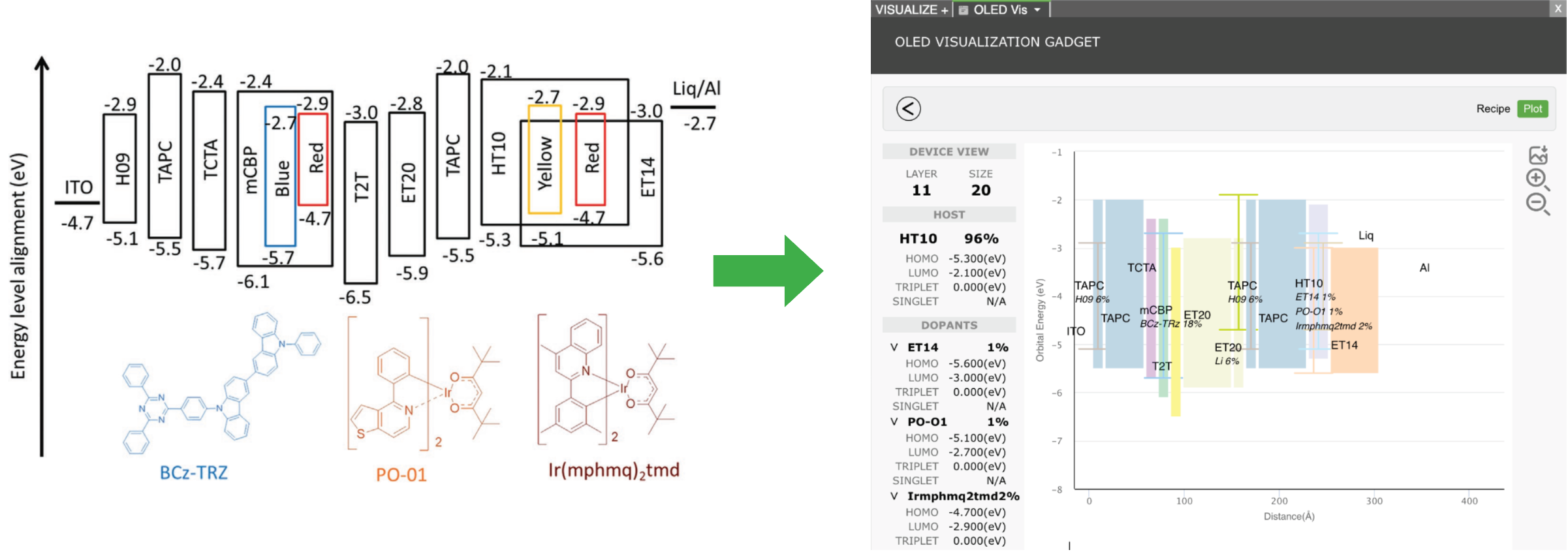
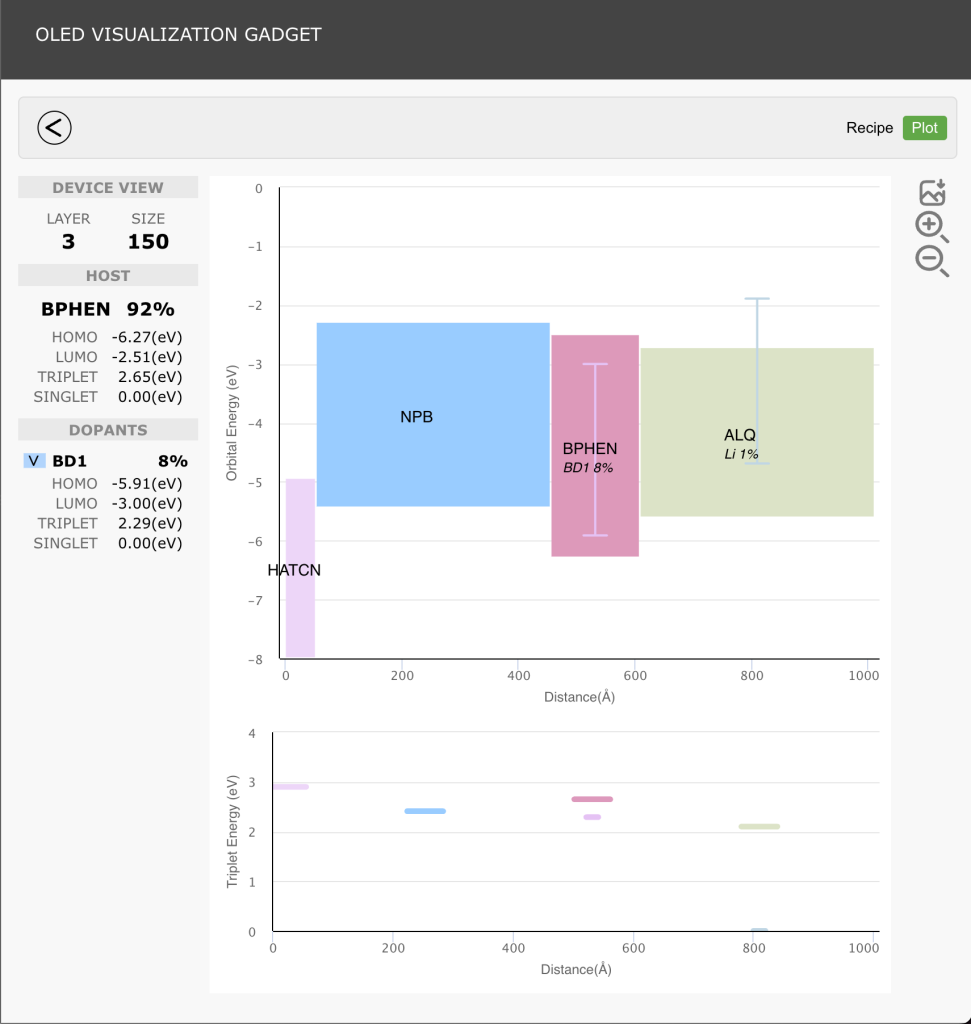
The rapidly evolving landscape of organic electronics demands innovative approaches for the design and development of materials, particularly for display applications. This presentation showcases the integration of machine learning and physics-based simulations for OLED material design and development. We explore the synergies between these computational techniques, unraveling the intricate thin-film morphology and electronic properties that underpin the performance of organic electronic materials. Viewed through a multidisciplinary lens, we navigate the complexities of OLEDs, uncovering key insights that drive the next generation of efficient and high-performance devices.
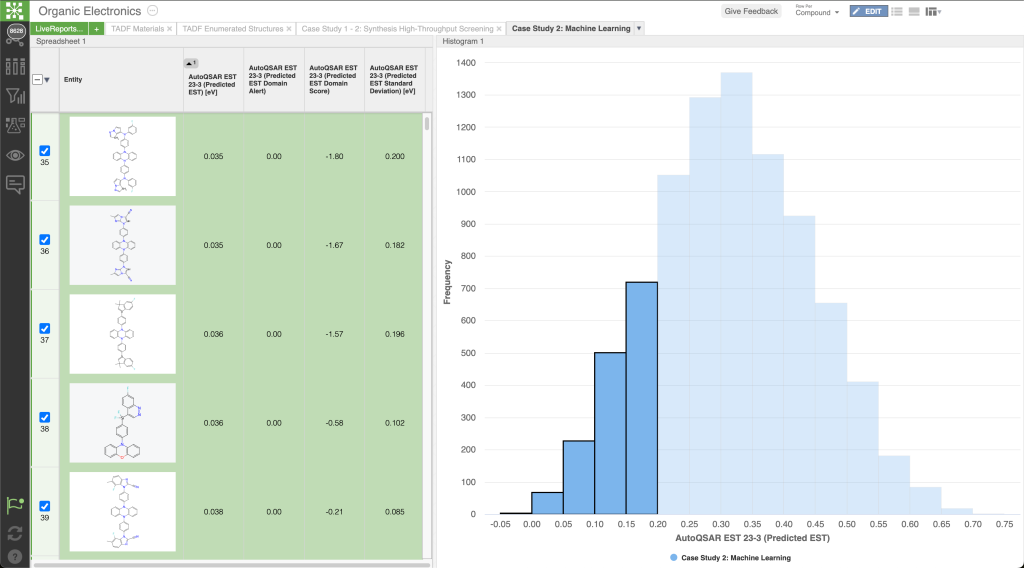
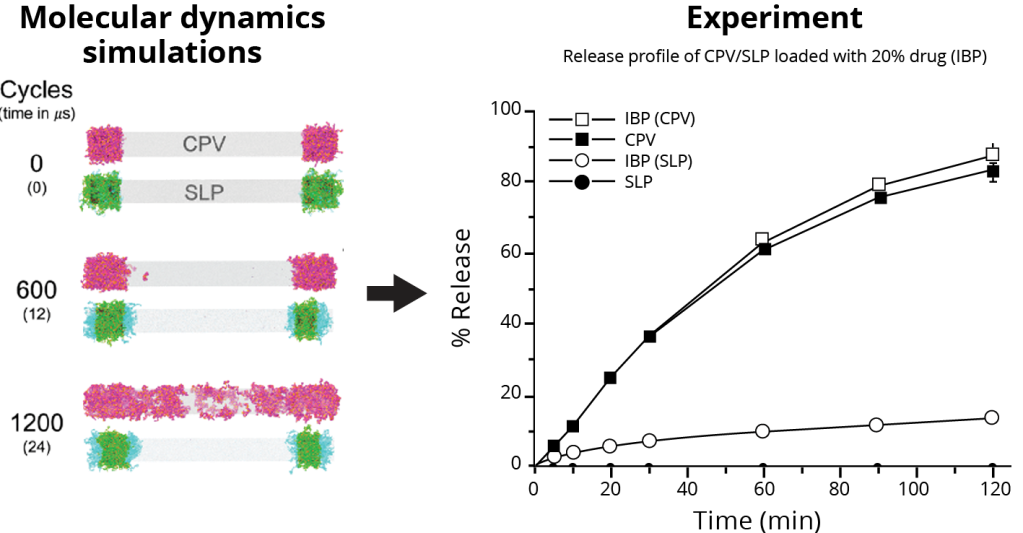
From understanding electronic transitions at the quantum level, morphology at the molecular level, to harnessing machine learning for accelerated material discovery, this presentation highlights the transformative impact of leveraging multiscale computational methodologies. We delve into several case studies, demonstrating how these computational tools empower researchers to predict, optimize, and tailor OLED materials for higher performance.
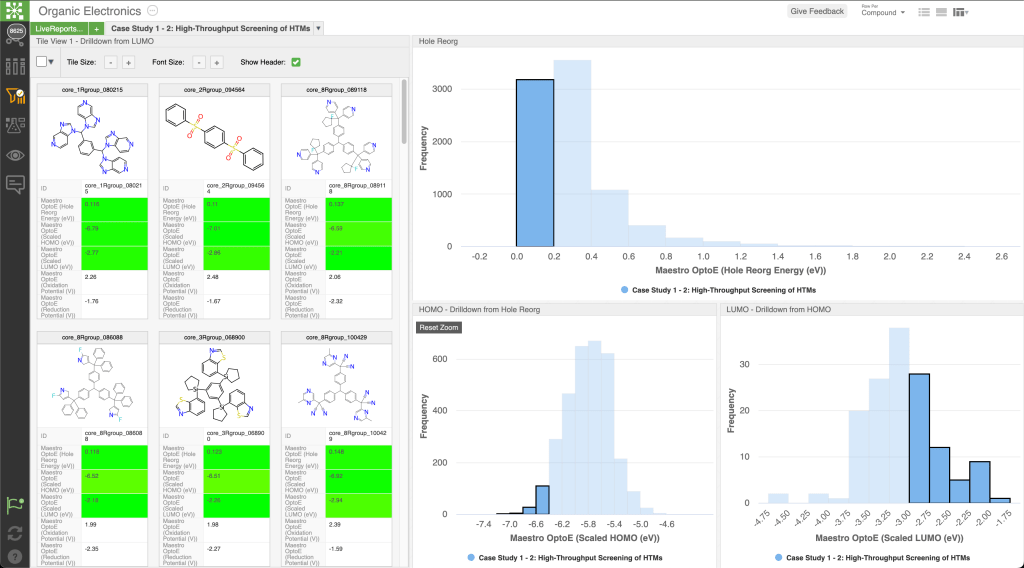
Addressing the challenge of managing extensive data in OLED design, we highlight Schrödinger’s informatics and collaboration tool, LiveDesign. This dynamic, cloud-native environment democratizes digital design processes, providing R&D teams with unified access to diverse tools such as physics-based modeling, advanced cheminformatics, and machine learning. LiveDesign streamlines collaboration and efficiency, overcoming limitations in expert availability and promoting innovation.
This presentation serves as a guide for researchers and industry professionals, pointing towards a future where computational insights drive the design and development of next-generation OLED materials.