Aggregation prediction with protein surface analyzer
Background
Biotherapeutics differ from small-molecule drugs not only in the size and complexity of the active ingredient but also in product manufacturing, storage, and delivery requirements. Production of antibodies, for instance, is an intricate process that relies on mammalian cell expression. Post-translational modifications occurring in mammalian cells mediate proper protein folding, multimerization, and secretion. Once produced, biologic drugs have to be kept at a high concentration under controlled storage conditions. Aggregation is one of the key risks that have to be managed throughout production and storage in order to maintain product safety, purity, and potency. Substantial time and monetary cost savings can be realized by early evaluation of relative aggregation tendencies of biomolecules and weeding out aggregation-prone candidates during the initial stages of a project. Unlike many current aggregation prediction methods that depend on primary sequence information, Schrödinger’s AggScore algorithm is entirely based on three-dimensional molecular structure. The structure-based approach of AggScore offers better accuracy and expanded general applicability compared to existing methods, in particular in situations when the differences of the compared structures are very subtle. In addition to surface-exposed hydrophobic regions, AggScore factors in the charge propensities of neighboring residues. The Protein Surface Analyzer and the AggScore metric are useful for 1) ranking and triaging proteins by aggregation propensity, 2) visualizing the distribution of aggregation‐prone regions on the surface of proteins and other biomolecules, and 3) reliably predicting the impact of residue mutation on aggregation behavior.
AggScore for Predicting Chromatography Retention Times of 137 Clinical-stage Antibodies
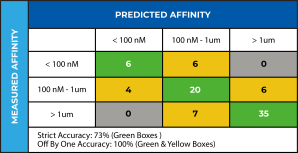
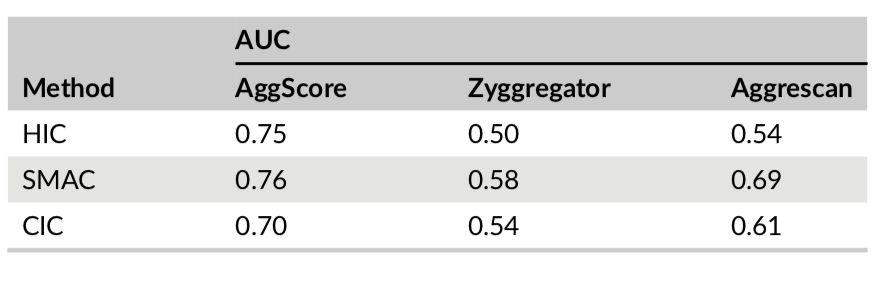
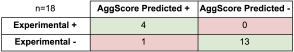
Three-dimensional structures of 137 clinical-stage mAbs were constructed using Schrödinger’s antibody modeling protocol. AggScore was then calculated for each of these structures and the scores compared with experimental retention times measured by Jian et al1 using three chromatographic methods – HIC, SMAC and CIC. The antibodies’ elution characteristics were classified as “early elution” (retention time < 10 min) or “delayed elution” (retention time ≥ 10 min). Prediction performance results summarized in Table 1 indicate that AggScore had the best AUC compared to two other commonly used computational approaches – Zyggregator and Aggrecan. The results demonstrate that AggScore’s domain of applicability includes antibodies.
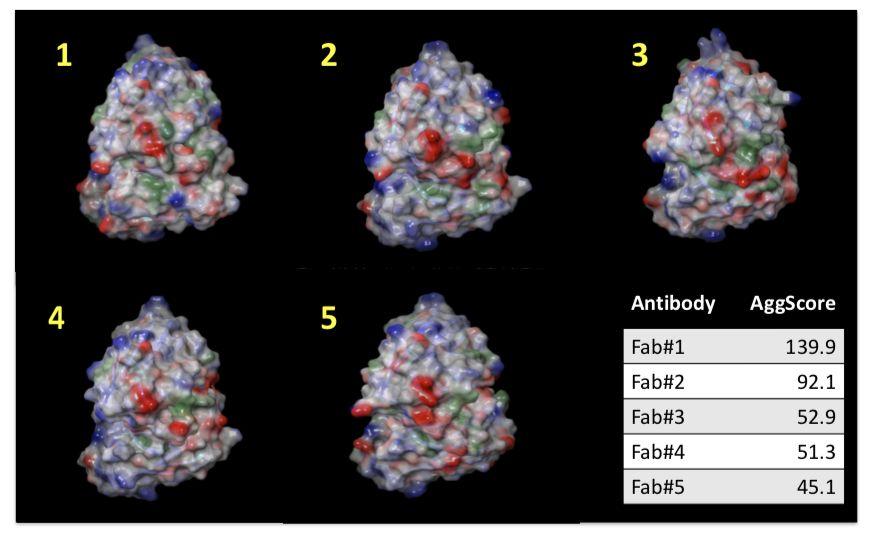
Mapping the Distribution of Aggregation‐prone Regions on the Surface of 5 Therapeutic Antibody Fv Fragments and Ranking them by Aggregation Propensity
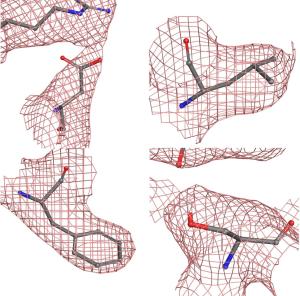
Patch properties were computed from molecular surfaces projected at the water-probe distance (1.4 Å) away from the vdW surface of the protein. The protein surface patch calculation determines three classes of surface patches based on the respective hydrophobic and hydrophilic surface potential values: hydrophobic (green), positive (blue) and negative(red). Input structures were refined prior to protein surface patch calculation. The system pH was set at the appropriate value and atom charges were assigned according to the OPLS3.0 force field. AggScore was calculated on the set of five antibody structures with known liabilities. The score was able to predict their aggregation propensities in perfect rank order (Figure 1).
Predicting the Impact of Residue Mutation on Aggregation Behavior
AggScore was used to predict aggregation propensities of several mutants of β-amyloid (Aβ). The predictions were compared with experimental data as shown in Table 2. The results indicate that:
- AggScore prediction matched experiment for eight out of nine single mutants (L17Q, L17E, F19S, I31N, I32S, I32V, L34P, V36E) of Aβ that showed reduced aggregation relative to the wild type (WT) as reported by Wurth et al5. The exception, A2S, is located at the n-terminal end of the protein, which does not contribute to AggScore.
- AggScore exactly reproduced the decreasing order of aggregation propensities of four mutants relative to wild type: WT > I41L > I41V >I41A > I41G.
- The prediction that three familial variants of Aβ, Dutch (E22Q), Iowa (D23N), and Italian (E22K) have higher aggregation propensity than the WT whereas the Flemish (A21G) has lower aggregation propensity than the WT is in good agreement with experimental studies performed by van Nostrand et al4 and Miravelle et al2.
Summary
Protein aggregation is a major impediment to the development of lead biomolecules into effective biotherapeutics. Schrödinger’s AggScore predicts aggregation propensities by taking into account residue contributions to charged and hydrophobic patch regions projected onto the surface of three-dimensional input structures. The method is well-suited to assist in identifying and mitigating aggregation issues in a variety of biologic product categories including antibodies, enzymes, and vaccine antigens.
References
-
Biophysical properties of the clinical-stage antibody landscape
-
Substitutions at codon 22 of Alzheimer’s Aβ peptide induce diverse conformational changes and apoptotic effects human cerebral endothelial cells
Miravalle et al. J Biol Chem. 2000, 275(35), 27110-27116
-
AggScore: Prediction of aggregation-prone regions in proteins based on the distribution of surface patches
Sankar et al. Proteins. 2018, 1–10
-
Pathogenic effects of D23N Iowa mutant amyloid β-protein
Van Nostrand et al. J Biol Chem. 2001, 276(35), 32860-32866
-
Mutations that reduce aggregation of the Alzheimer’s Aβ42 peptide: an unbiased search for the sequence determinants of Aβ amyloidogenesis
Wurth et al. J Mol Biol. 2002, 319(5), 1279-1290