
SEPT 29, 2021
A Molecular-Level Examination of Amorphous Solid Dispersion Formulation Dissolution
Speakers
Samuel Kyeremateng, Atif Faiz-Afzal, John Shelley
AbbVie and Schrödinger
Samuel Kyeremateng, Atif Faiz-Afzal, John Shelley
AbbVie and Schrödinger
Atif Faiz-Afzal
Principal Scientist, Schrödinger

Chris Brown, Executive Director at Schrödinger
John Shelley, Fellow at Schrödinger
Virtual screening has proven to be a very successful approach for finding ligand hits and assisting lead optimization in structure-based drug discovery projects. By docking a large library of compounds into one or more high-resolution structures of the target receptor, fewer compounds typically need to be experimentally screened to identify prospective lead optimization candidates. Beyond identifying small molecules likely to bind well to a protein target, docking methods are used in a variety of context such as polypeptide and macrocycle pose prediction, predicting protein-ligand complex geometries, and preparing congeneric series for binding affinity prediction with methods such as Free Energy Perturbation or MM-GBSA. Moving beyond the rigid receptor approximation common in structure-based virtual screening, the Induced Fit docking protocol predicts the effect of ligand docking on protein structure.
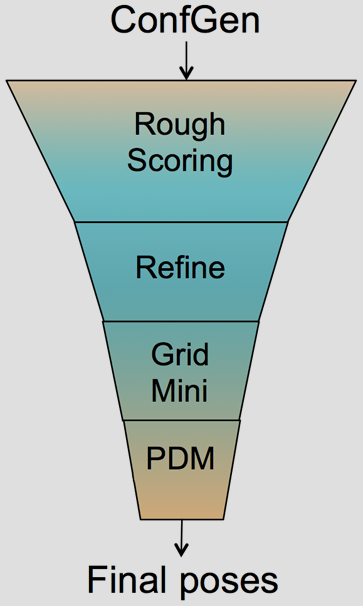
The Glide HTVS, SP and XP docking methodologies have previously been described in detail.1-3 Glide HTVS and SP use a series of hierarchical filters to search for possible locations of the ligand in the binding-site region of a receptor. The shape and properties of the receptor are represented on a grid by different sets of fields that provide progressively more accurate scoring of the ligand pose. Exhaustive enumeration of ligand torsions generates a collection of ligand conformations that are examined during the docking process. Given these ligand conformations, initial screens are deterministically performed over the entire phase space available to the ligand to locate promising ligand poses. From poses selected by initial screening, the ligand is refined in torsional space in the field of the receptor using OPLS34 (Glide SP & XP) or OPLS2005 (GLIDE HTVS) with a distance-dependent dielectric model. Finally, a small number of poses are minimized within the field of the receptor with full ligand flexibility (post-docking minimization or PDM). This process is referred to as the docking funnel as illustrated in Figure 1.
Glide HTVS can dock compounds at a rate of about 2 seconds/compound and trades sampling breath for higher speeds. Glide SP performs exhaustive sampling and is the recommended balance between speed and accuracy, requiring about 10 seconds/compound. Glide XP employs an anchor-and-grow sampling approach and a different functional form for GlideScore. It can dock compounds at a rate of about 2 minutes/compound. These three docking modes provide an array of options in the balance of speed vs. accuracy for most situations.
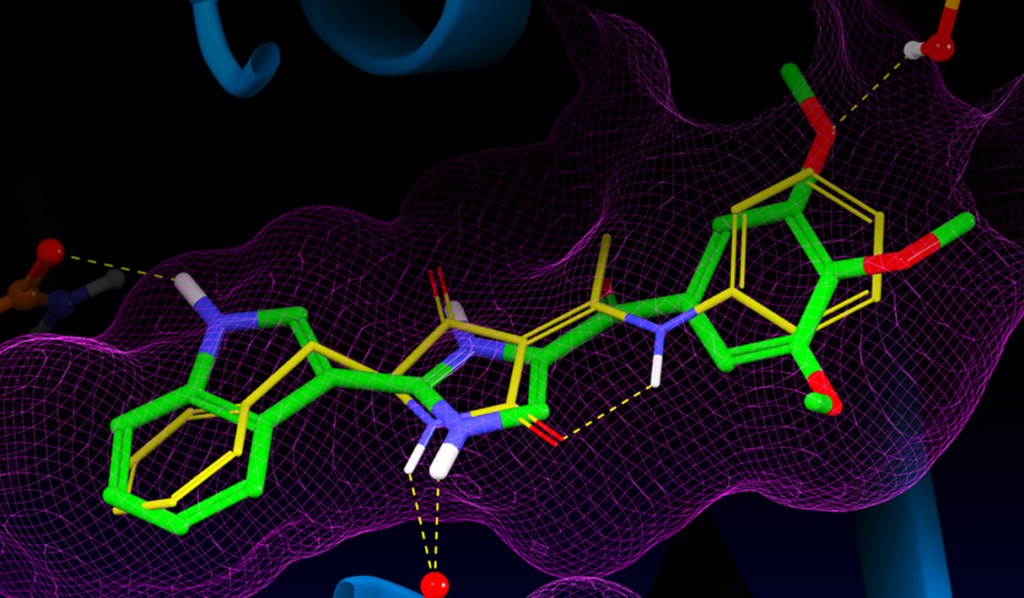
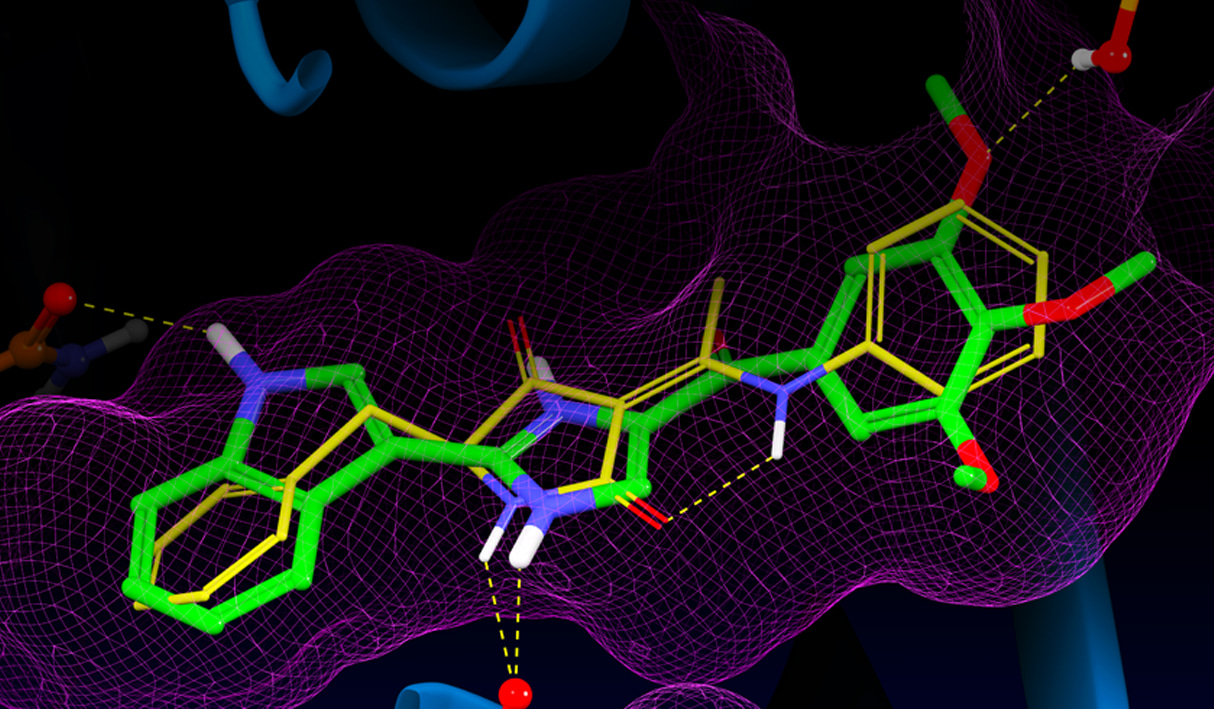
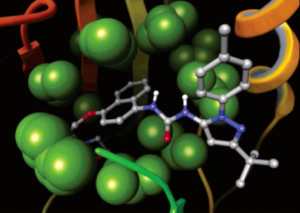
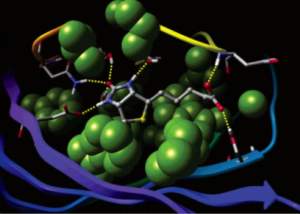
Glide uses the Emodel1 scoring function to select between protein-ligand complexes of a given ligand and the GlideScore2 function to rank-order compounds to separate compounds that bind strongly (actives) from those that don’t (inactives). The Emodel scoring function is primarily defined by the protein-ligand coulomb-vdW energy with a small contribution from GlideScore. GlideScore is an empirical scoring function designed to maximize separation of compounds with strong binding affinity from those with little to no binding ability. As an empirical scoring function it is comprised of terms that account for the physics of the binding process including a lipophilic-lipophilic term, hydrogen bond terms, a rotatable bond penalty, and contributions from protein-ligand coulomb-vdW energies. In addition to these terms, GlideScore includes terms to account for hydrophobic enclosure3, which is the displacement of water molecules by a ligand from areas with many proximal lipophilic protein atoms. Particularly beneficial to binding is the formation of one or more protein-ligand hydrogen bonds within regions of hydrophobic enclosure as illustrated in Figures 2 and 3.
The first Glide article1 is the 2nd most cited paper ever in the Journal of Medicinal Chemistry (as of Feb. 2016) with many references citing its application in drug discovery projects. Glide’s ability to consistently identify hits for lead optimization and guide understanding of the key interactions and desolvation effects impacting protein-ligand binding have helped drive its widespread adoption. A pre-requisite to obtaining the highest-quality docking results is to use protein and ligand structures prepared using best-practices methods by the Protein Preparation Wizard and LigPrep. As part of an unbiased comparison of docking methods, we have evaluated Glide SP for binding mode prediction using the Astex5 set of self-docking tests, and enrichment in a virtual screening context using the DUD dataset6. The datasets, evaluation metrics, and experiments performed were prescribed by conditions of the Docking and Scoring Symposium at the 241st ACS Meeting in Anaheim.7
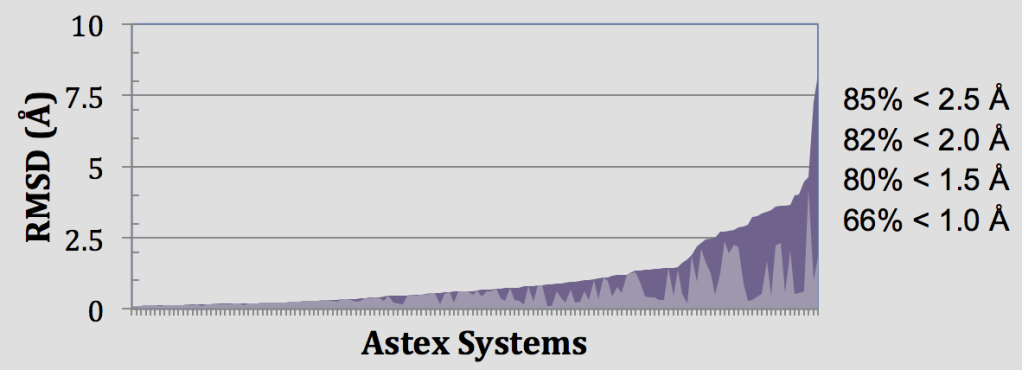
In these retrospective experiments, Glide SP reproduces crystal complex geometries in 85% of Astex cases with < 2.5 Å RMSD as shown in Figure 4. Glide is also effective in enriching screened databases in known actives, beating random selection in 97% of DUD targets and giving an average AUC of 0.80 across all 39 target systems. Early enrichment performance shows on average 12%, 25%, and 34% of known actives are recovered in screening the top-ranked 0.1%, 1%, and 2% of recovered decoys. This enrichment is particularly impressive given the structural similarity between active and decoy ligands in the DUD dataset8 and that docking a ligand with SP Glide takes only about 10 seconds using modern hardware.
| ROC(0.1%) | ROC(1%) | ROC(2%) | AUC | |
|---|---|---|---|---|
| Best-Practices Structures | ||||
| Average | 0.12 | 0.25 | 0.34 | 0.80 |
| Median | 0.06 | 0.19 | 0.27 | 0.14 |
| Std. Dev. | 0.17 | 0.22 | 0.24 | 0.14 |
| Maximum | 0.82 | 0.88 | 0.88 | 0.98 |
Glide’s wide range of constraints enable one to “stay close to experiment“ which has been reported as a common theme in pharmaceutical projects at Roche that were positively impacted by computational chemistry.9 Hydrogen bond constraints require formation of a hydrogen bond to particular functional groups on the receptor, positional constraints can enforce docked poses to place particular chemistry in a volume of space relative to the receptor, metal constraints enforce coordination geometries. Core constraints are particularly valuable for enforcing expected binding modes of congeneric series in preparing docked poses for binding affinity prediction via MM-GBSA or Free Energy Perturbation.
Efficient sampling of ring conformations is essential to reproduce experimental binding modes in docking experiments, particularly for macrocycles where there can be hundreds of low energy conformations. Glide relies on an extensive database of ring conformations to sample low-energy states and otherwise does not sample macrocycle ring systems. Generating ring templates for macrocycles considerably improves native pose prediction.
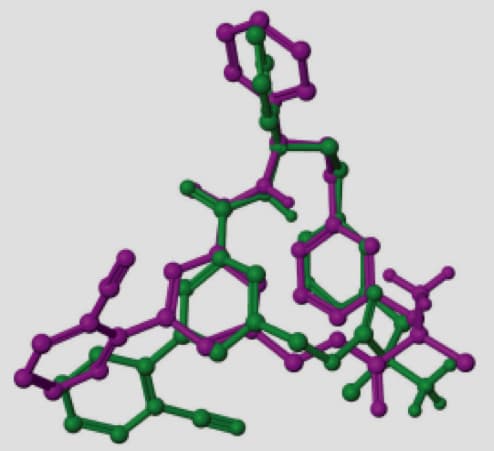
A representative example is the macrocycle from 2QKZ shown in this figure where the native ligand buries the macrocycle ring into a mostly enclosed pocket. To bind in this pocket requires a particular ring conformation that forms key interactions to a ligand carbonyl and protonated amine moieties. Using ring templates for this macrocycle the docked pose has a low RMSD of 0.22 Å. Without the macrocycle ring templates, even with a visually similar ring conformation of RMSD 0.71 Å for macrocycle ring atoms, Glide positions the ring outside the pocket leading to a very large RMSD of 10.23 Å.
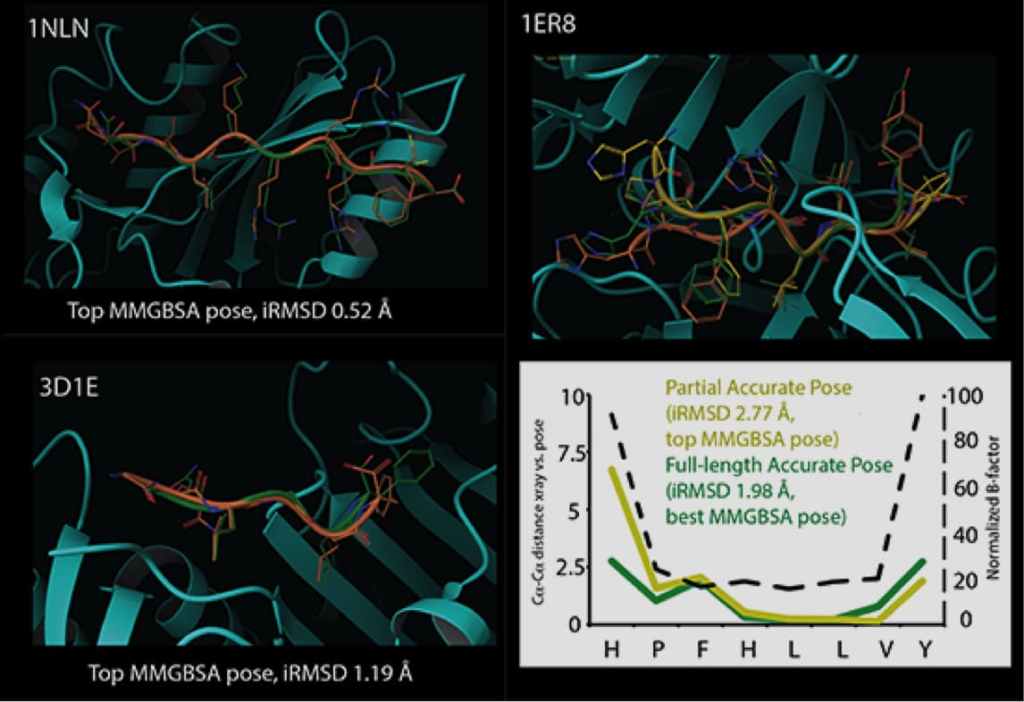
The increased use of peptides and peptidomimetics as drugs has created a need for the accurate prediction of binding complex geometries of polypeptides to receptors. The Glide SP-peptide mode10 was developed to address this need as default application of Glide to such ligands results in a successful docking for 24% of the peptides in a benchmark set. To improve binding mode prediction, the Glide sampling algorithm was modified by systematically changing all parameters that control how peptide conformations make it through the Glide funnel. Applying the optimal combination of these parameters increased pose prediction accuracy to 41%. In addition, we leveraged the previously reported sensitivity of Glide to input conformations11 and increased sampling by running multiple independent Glide jobs and pooling the results. This increased accuracy of pose prediction to 53%. Finally, by applying MM-GBSA scoring to each of the predicted poses, further boosting the accuracy to 58%. This approaches accuracy of brute-force methods like FlexPepDock (63% for this dataset) at a fraction of the computational cost (24 CPU hours for the Glide protocol).
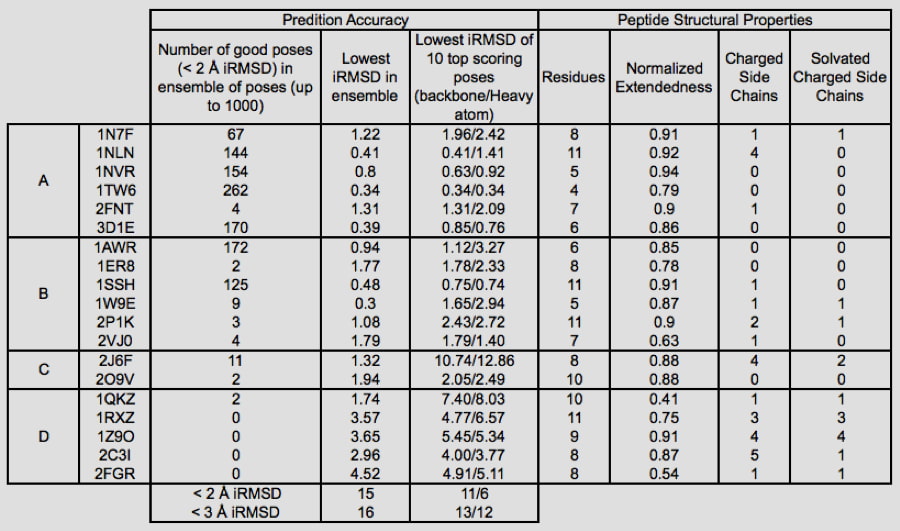
Glide can dock ligands with up to 300 atoms and 50 rotatable bonds. Since many peptide ligands may exceed these limits, this effectively limits docking application to peptides of around 11 residues or less. In some cases, it is necessary to freeze the amide bonds in their trans configuration to dock peptides of longer lengths. Strong correlations are found between structural features of peptides and their ability to be docked accurately. For example, we found that reduced peptide length, increased extendedness, and small number of ionizable groups increased the probability that a peptide was docked correctly. In addition, we noted a strong correlation between the quality of prediction for segments of the peptides with low B-factors, while the high B-factor (typically the N- and/or C- termini) would have significant deviation from the crystal structure, suggesting that these terminal predictions might actually be accessible states in solution.
The active site geometry of a protein complex depends heavily upon conformational changes induced by the bound ligand. However, resolving the crystallographic structure of a protein-ligand complex requires a substantial investment of time, and is frequently infeasible or impossible. Schrödinger’s Induced Fit (IFD) protocol solves this problem by using Glide and Prime to exhaustively consider possible binding modes and the associated conformational changes within receptor active sites. The unique procedure allows chemists to quickly predict active site geometries with minimal expense, even for systems as challenging as hERG homology models.
The Induced Fit protocol begins by docking the active ligand with Glide. In order to generate a diverse ensemble of ligand poses, the procedure uses reduced van der Waals radii and an increased Coulomb-vdW cutoff, and can temporarily remove highly flexible side chains during the docking step. For each pose, a Prime structure prediction is then used to accommodate the ligand by reorienting nearby side chains. These residues and the ligand are then minimized. Finally, each ligand is re docked into its corresponding low energy protein structures and the resulting complexes are ranked according to a scoring function combining GlideScore and Prime energies.
The Induced Fit methodology has been thoroughly refined in real-world research applications, and is readily used by novice and expert modelers alike. As shown in Table 3 binding pose prediction of Induced Fit for a range of targets where protein conformational changes are necessary for binding is very good. In addition to default settings suitable for a wide range of systems, the Induced Fit interface features advanced options that can be customized to solve more challenging cases. Calculations can be completed in a matter of hours on a desktop machine, or in as few as 30 minutes when distributed across multiple processors.
| RMSD of Top-Ranked Poses Returned by Induced Fit
|
||||
|---|---|---|---|---|
| Target | Receptor | PDB source of ligand structure | Docking RMSD before Induced Fit (Å) |
Ligand RMSD after Induced,Fit (Å)
|
| Aldose reductase | 2acr | 1ah3 | 6.5 | 0.9 |
| Antibody DB3 | 1dba | 1dbb | 7.6 | 0.3 |
| CDK2 | 1buh | 1dm2 | 6.4 | 1.1 |
| CDK2 | 1dm2 | 1aq1 | 6.2 | 0.8 |
| CDK2 | 1aq1 | 1dm2 | 0.6 | 0.8 |
| COX-2 | 3pgh | 1cx2 | 11.1 | 1 |
| COX-2 | 1cx2 | 3pgh | 6.6 | 1.0 (0.51) |
| Estrogen receptor | 1err | 3ert | 5.3 | 1 |
| Estrogen receptor | 3ert | 1err | 2.3 | 1.4 (1.02) |
| Factor Xa | 1ksn | 1xka | 9.3 | 1.5 |
| Factor Xa2 | 1xka | 1ksn | 5.3 | 1.5 |
| HIV-RT | 1rth | 1c1c | 2.5 | 1.3 |
| HIV-RT | 1c1c | 1rth | 12 | 2.5 |
| Neuraminidase | 1nsc | 1a4q | 3.9 | 0.8 |
| Neuraminidase | 1a4q | 1nsc | 1 | 1.7 |
| PPAR – gamma | 1fm9 | 2prg | 9.1 | 1.8 (0.43) |
| PPAR – gamma | 2prg | 1fm9 | 9.8 | 3.0 (1.54) |
| Thermolysin | 1kr6 | 1kjo | 1.1 | 1.3 |
| Thermolysin | 1kjo | 1kr6 | 3.5 | 3.2 (1.65) |
| Thymidine Kinase | 1kim | 1ki4 | 4.7 | 0.4 |
| Thymidine Kinase | 1ki4 | 1kim | 0.5 | 1.2 |
In recent years, there has been a growing interest in the design of drugs forming a covalent bond with the target protein, with nearly 30% of the marketed drugs targeting enzymes known to act by covalent inhibition12. Covalent interaction with the target protein has the benefit of prolonged duration of the biological effect and potential for improved selectivity. Examples of proteins that have been targeted by covalent drugs include serine penicillin-binding proteins (PBPs) which bind to β lactams and β-lactone antibiotics, cysteine proteases such as Cathepsin B, K, and S, which are covalently modified by vinyl sulfones, epoxides, isothiazolones, and ketoamides, as well as other chemical groups, and hepatitis C virus (HCV) protease, which covalently binds the ketoamide groups of boceprevir and telaprevir.
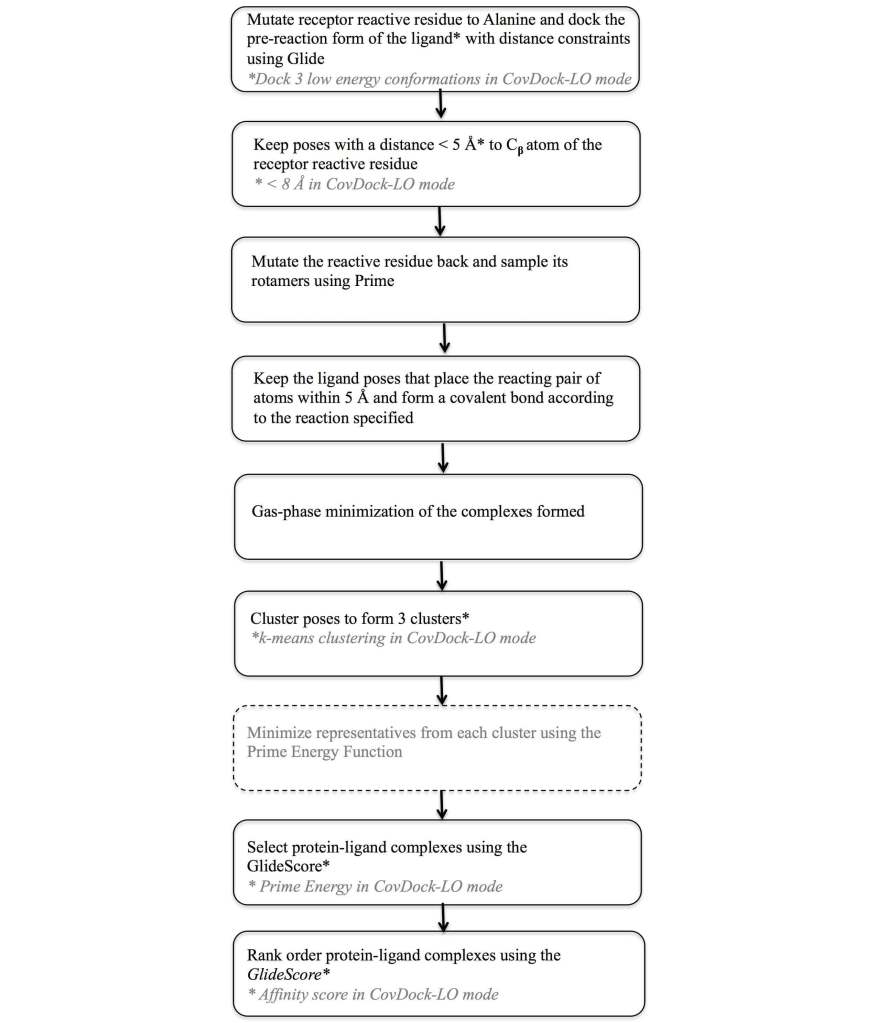
To address this need a covalent docking protocol, CovDock13, 14 was developed that yields good accuracy in binding mode prediction and has automatic detection of reactive atoms using SMARTS patterns. Using a workflow that combines Glide docking and Prime optimization, the CovDock-VS (virtual screening) and CovDock-LO (lead optimization) modes enable application of covalent docking in large-scale virtual screening or lead optimization projects, respectively. The workflow for these two approaches is shown in Figure 7. Thanks largely to reduced sampling, the CovDock-VS mode is 10 to 40 times faster then CovDock-LO mode.
Docking accuracy for CovDock is good with an average RMSD of 1.9Å for CovDock-VS and 1.5 Å for CovDock-LO in a retrospective native docking experiment with results shown in Table 4. In combination with interaction filters (in these cases, H-bond constraints), CovDock-VS has also demonstrated excellent enrichment in retrospective experiments as shown in Table 5. In a recent test it has been shown to retrieve 71%, 72%, and 77% of the known actives for Cathepsin K, HCV NS3 protease, and EGFR within 5% of a decoy library, respectively. Even in cases where interaction filters could not be used, as for XPO1, CovDock-VS was still able to retrieve 95% of known actives within the top 30% of decoy compounds, with 33% of known compounds found in the top 1% of decoys.
| CovDock-VS | CovDock-LO | ||
|---|---|---|---|
| Cathepsin K actives | |||
| 1YT7-ligandc | 2.68 | 0.68 | |
| 2BDL-ligand | 2.48 | 2.89a | |
| 1TU6-ligand | 1.43 | 3.18 | |
| HCV NS3 protease actives | |||
| 3KNX-ligand | 2.89a | 2.45 | |
| 2GVF-ligand | 1.66 | 1.51 | |
| 3LON-ligand | 6.69a | 1.81 | |
| 2OC7-ligand | 1.38 | 1.12 | |
| 2F9U-ligand# | 1.19 | 1.30 | |
| 2OBO-ligand | 1.29 | 0.98 | |
| 2OC8-ligand | 1.55 | 1.04 | |
| 2A4Q-ligand | 2.51 | 2.28 | |
| 2OC0-ligand | 3.03 | 0.88 | |
| 3LOX-ligand | 2.07 | 1.72 | |
| 2OC1-ligand | 3.75a | 3.14 | |
| EGFR actives | |||
| 4I24-ligand (Dacomitinib) | 1.46 | 0.85 | |
| 4G5J-ligand (Afatinib) | 1.81 | 0.86 | |
| 3IKA-ligand (WZ4002) | 2.53a | 1.67 | |
| 2JIV-ligand (Neratinib) | 6.54a | 2.41a | |
| 2J5F-ligand (34-JAB) | 1.57 | 1.20 | |
| XPO1 actives | |||
| 4GMX-ligand (KPT-185)c | 2.19b | 0.67 | |
| KPT-276 (not submitted to PDB)c | 2.54a | 1.07 | |
| Mean | 1.87 | 1.50 | |
| EF1% | EF10% | BEDROC (α = 20) | |
|---|---|---|---|
| HCV NS3 protease | 52 | 7 | 0.70 |
| Cathepsin K | 9 | 8 | 0.48 |
| EGFR | 46 | 8 | 0.65 |
| XPO1 | 33 | 7 | 0.52 |
J. Med. Chem., 47, 1739-49 (2004).
J. Med. Chem., 47, 1750-59 (2004).
J. Med. Chem., 49, 6177-96 (2006).
J. Chem. Theory Comput., 12(1), 281-296 (2016).
J. Med. Chem., 50, 726-741 (2007).
J. Med. Chem., 49, 6789-6801 (2006).
J. Comput. Aided Mol. Des., 26, 787-99 (2012).
J. Med. Chem., 55, 6582-94 (2012).
J. Med. Chem., 9(9), 4087–4102 (2016).
J. Chem. Inf. Model., 53, 1689-99 (2013).
J. Chem. Inf. Model., 52(3), 724–738 (2012).
Biochemistry, 44, 5561-71 (2015).
J. Chem. Inf. Model., 54, 1941-50 (2014).
J. Chem. Inf. Model., 54, 1932-40 (2014).
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Panasonic scientists in collaboration with Schrödinger researchers published a paper in The Journal of Physical Chemistry, Massive Theoretical Screen of Hole Conducting Organic Materials in the Heteroacene Family by Using a Cloud-Computing Environment, about performing a large-scale theoretical screen of potential organic electronic materials in a cloud computing environment to determine molecules with optimal hole mobility properties. The performance of organic semiconductor devices depends crucially on the movement of charge through the weakly conducting materials composing the device. Positive charge carriers and negative charge carriers are holes and electrons, respectively. Charge transport can occur by hopping from molecule to molecule through the solid, modeled using atomic scale simulation techniques in the Schrödinger platform. The focus in this work was hole mobility in organic semiconductors.
Panasonic was interested in exploring structures with fused furans, thiophenes and selenophenes. These compounds are interesting for their use as organic semiconductors for a variety of applications. In particular, Panasonic is interested in making printable RF-ID (radio frequency identification) tags that require organic semiconducting materials with charge mobilities higher than currently available compounds. These materials could be used in housing, retail and warehouse management. Noncontact checkout and inventory would further enable future retail transaction technology. For example, a current Panasonic product, called ‘Reji-Robo’ , is a new automated checkout system that cuts labor cost by 10% while speeding up the process for customers.
In this work, Schrödinger created 7,032,432 compounds through structural enumeration as the basis of chemical space for high-performance hole conducting materials. 250,000 of the initial set were randomly selected to perform density functional theory (DFT) calculations of hole reorganization energies. Because of the great amount of compounds to analyze, the team decided to perform the calculations in a cloud computing environment to drastically increase the throughput of calculations.
Leveraging Google cloud computing technology, 3.6M DFT calculations were run to predict hole reorganization energies of the 250,000 compounds—this was completed in just 16 days.

Alexander Goldberg, Senior Principal Scientist within Schrödinger’s Materials Science Group, explains the paper’s key findings:
Can you explain why this work is important?
Obtaining organic materials with improved mobility will help advance the field of organic electronics. Because organic electronic materials are a low-cost, highly flexible, easily processed, lightweight, and sustainable solution there has been a surge in both interest and investment in this field. The global organic semiconductor market alone is expected to grow to $179.4 Billion by 2024, up 22.4% from 2019.1
Although performance of organic material-based devices has improved to the point that it is now challenging inorganic materials for key applications, fundamental research is still needed for further development since the underlying physics is still unclear and working principles need to be clarified. We can utilize the Schrödinger platform to advance this research and enable design of high-quality, next-generation organic electronic materials.
Explain what this paper means. Why is it so exciting?
Performing a screening of this size in this amount of time wasn’t possible even just a few years ago. With the improvements seen in cloud computing, we’ve been able to increase the scale of using atomic scale simulation to screen chemical space drastically. When you consider that screening this number of compounds would take 100 days using a typical 200 CPU HPC resource, you can see just how much faster it is using cloud computing. If you were to screen this number using traditional experimental methods, it would take somewhere around 5,000 years.
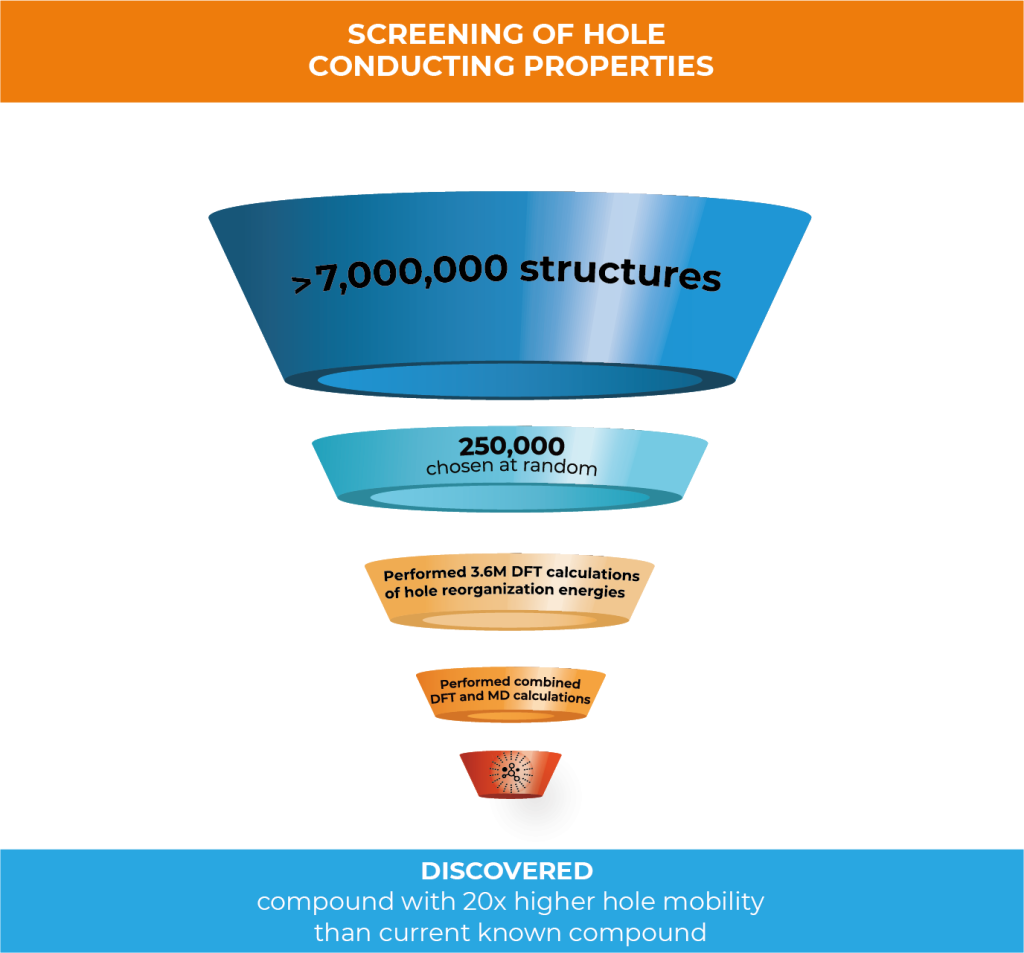
In this project, you evaluated the suitability of 250,000 structures as hole transport materials from a chemical library of 7,032,432 structures to find structures with the most optimal hole mobility. Can you describe the process you used?
250,000 structures were randomly selected to perform density functional theory (DFT) calculations of hole reorganization energies. Then, the hole mobilities of compounds with the lowest 130 reorganization energy were further processed by applying combined DFT and molecular dynamics (MD) methods. By using this method, we were able to identify structures with a predicted hole mobility that is 20 times higher than dinaphthothienophene (DNTT)—a state of the art compound with one of the highest experimental mobilities observed.2
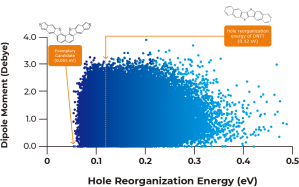
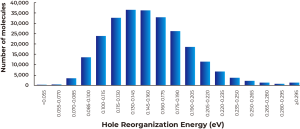
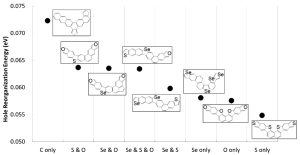
As described in Figure 1A above, calculated dipole moments are plotted against hole reorganization energies for the 250,000 compounds. Good charge transport materials should have small dipole moments to avoid charge trapping in the solid state. The hole reorganization energies range from 0.055 to 0.50 eV. From there, we narrowed down to 17,000 compounds that had hole reorganization energy values below 0.1 eV, as shown in Figure 1b. When you compare that to DNTT which has a hole reorganization energy of 0.12 eV, these 17,000 compounds could all be judged as promising candidates for improved hole transport materials.
Figure 2 shows the minimum calculated hole reorganization energy in every considered topological compound together with their corresponding chemical structure.
Was there a stepwise selection carried out to filter for high mobility candidates?
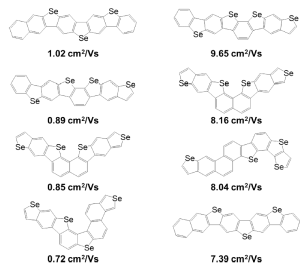
Yes. For example, because the cost of calculating mobility is much higher than calculating reorganization energy, after the massively parallel DFT calculations on the cloud, we selected 130 compounds with the lowest hole reorganization energy for further calculations to obtain estimated condensed phase hole mobility. By calculating 130 compounds we were able to rank which ones had the highest hole mobilities. Figure 3 (below) shows the chemical structures of compounds with the top four highest hole mobilities from that group. Two methods based on different approximations were used to compute mobility in ranking the candidates. Percolation theory is based on a random walk approach, whereas energy disorder theory is based on the distribution of energy states in the solid.
Are there other examples of how Schrödinger uses cloud computing?
Schrödinger utilizes cloud computing not only in Materials Design, but also in our Drug Discovery and Biologics research. Cloud computing enables us to screen vast areas of chemical space in a fraction of the time it would take with on-premise computing resources.
Schrödinger has a history of breaking records for virtual screening in the cloud. In 2013, our materials science team partnered with Cycle Computing, creating a 156,000+ core cloud computing run that was dubbed the Megarun.3 It used virtual machines across eight regions of Amazon Web Servcie’s public cloud around the world for a total of 18 hours and a total cost of just $33,000. It was the largest and fastest cloud computing run at that time in history. When you look at that milestone compared to this screening, you can see how quickly cloud computing technology is advancing.
Earlier this year, Schrödinger partnered with Google Cloud to accelerate our research. With nearly limitless processing power on demand, we are able to explore a larger area of chemical space and model vastly more compounds in a fraction of the time it would take using on-premise computing.
Google also donated resources to advance our work in the fight against COVID-19. Schrödinger is part of a philanthropic initiative with leading biopharma companies from around the world to develop antiviral therapeutics. Google Cloud is providing 16 million hours of GPU time as part of this collaboration. It’s equivalent to accessing the world’s fastest supercomputers.
What’s next?
Our next step is to continue our close collaboration with Panasonic and use the results from this study to enable machine learning techniques to determine structure-property relationships and further screen organic semiconductor design space for high mobility application.
Any final thoughts?
Cloud computing has revolutionized how we do research. Atomic scale simulation is already making a tremendous impact on materials R&D, lowering cost, shortening timelines, reducing risk and driving innovation. Cloud provides essentially, “limitless” resources. We no longer have to limit our pool of candidates to specific libraries. Being able to access the expanse of chemical space means we have a better chance of finding the best candidates with the most optimal properties. And, being able to do it faster means we are able to get results quicker that inform what next steps are needed. It’s an exciting time to be working in materials research.

Ph.D., earned his Ph.D. in Chemical Physics from Tel Aviv University in Israel
Dr. Goldberg has been with Schrödinger for 8 years. His personal research focused on absorption, emission and IR spectroscopy of small clusters. He was also involved in the development of photochemical reactions of organic molecules to be employed in optoelectronic devices as well as finding materials for hydrogen storage in fuel cells and Li battery technology. His research was based on molecular simulations at different length- and time-scales using computational methods of quantum mechanics, classical physics and mesoscale modeling.
Market Research Future; Published September 2020
Brandon Butler; Network World; November 12, 2013

Schrödinger, Inc, a company founded on the idea that computers can dramatically accelerate the discovery of better (more efficacious and less toxic) drugs, has realized this vision by making breakthroughs in scientific algorithms and by leveraging Moore’s law. The technology is at an inflection point such that the accuracy of biomolecular simulations is now consistently high enough to be used to truly drive preclinical drug discovery projects. This promises to transform the drug discovery industry, in the same way that computers have transformed nearly all other industries ranging from movie making to manufacturing of airplanes. The strategic inflection point in nanoscale simulation presents investors with an opportunity to ride a wave of rapid innovation and exceptional value creation.
At various times over the past 30 years, computer technology has been advanced as a means of dramatically increasing the efficiency of developing new drugs. The cost of computing has declined by a factor of 2 roughly every 18-24 months, in accordance with Moore’s law, for more than 50 years. In contrast, and somewhat alarmingly, the increase in the average cost to develop a new, FDA approved drug has increased steadily over the past 15 years, and now stands at approximately $1 billion. This phenomenon, which has been given the name Eroom’s law (Moore’s law spelled backwards), threatens not only the viability of major pharmaceutical and biotechnology companies, but the ability of our society to continue the vast improvements made in health care in the 20th century at an affordable cost. The large increase in costs has limited the rate at which new life-saving medicines are produced.
A recent analysis of 10 years’ worth of small-molecule drug discovery projects from four large companies – Pfizer, AstraZeneca, Eli Lilly, and GlaxoSmithKline – reveals a key cause of failure: poor properties such as absorption, distribution, metabolism, excretion, and/or toxicity (collectively referred to as ADMET properties) of the drug candidates. Even at the preclinical stage (prior to human trials), only 14% of 400 compounds declared as “development candidates” successfully passed the toxicity animal testing required prior to compounds entering into human trials. In Phase 1 clinical trials, ADMET problems accounted for more than 50% of failure to progress to Phase 2. These attrition rates have many possible specific causes, but one thing is clear; despite major advances in chemical synthesis, it still costs a lot of money to synthesize a drug candidate (on average $5,000) – so only a few thousand compounds are typically made over the course of a project. And despite the availability of atomic resolution structures for many important drug targets, it is still extraordinarily difficult to design just the right molecule – so a few thousand compounds may not be enough to achieve success.
The industry has recognized for a long time that technological breakthroughs are needed to substantively increase the success probabilities of small molecule drug discovery projects. Many such breakthroughs have been claimed over the past 30 years: structure-based drug design, high-throughput screening, combinatorial chemistry, systems biology, and genomics are some of the most prominent examples. While these techniques are currently used in the pharmaceutical and biotechnology industries, none has proven to be a panacea in struggling with the combination of higher safety and efficacy requirements for new drugs.
As mentioned above, computer technology has been advanced as a means of dramatically increasing the efficiency of developing new drugs. Computers are in fact routinely used by drug discovery teams to store and retrieve chemical and biological information, monitor the progress of projects, and enable graphical visualization of the structures of drug candidates and the proteins they target. However, these applications do not exploit the massive increases in computing power reflected in Moore’s law. The question is whether there is any computational approach in which large scale computing capability can be used inexpensively and efficaciously to select better drug candidates. As will become evident below, the answer is a resounding yes.
The focus here will be on small-molecule drugs (molecules with typically fewer than 100 atoms), which currently constitute ~75% of FDA approvals, but similar questions can be asked about biologics (e.g., antibody therapeutics) as well. Small-molecule drug discovery is a search for the most valuable substances known to man; those that can ameliorate or cure debilitating disease. At the dawn of the pharmaceutical era, Paul Ehrlich, who discovered one of the first effective drugs produced by chemical synthesis, envisioned the existence of a “magic bullet” – a molecule perfectly suited to its prescribed medical function. This concept still drives much of the $50B per year spent on pharmaceutical research.
How can such a molecule be identified? In a typical small-molecule drug discovery project, biological research first identifies a “target”; a protein in the body, or in a pathogen, that one would like to block (or in some cases activate) with the drug molecule. For example, current standard of care AIDS medication, which has transformed AIDS infection from a death sentence to a chronic condition, consists of a “cocktail” of three different molecules, each of which blocks or inhibits a protein made by the AIDS virus, which is necessary for it to reproduce itself. The targets for AIDS were identified roughly 30 years ago; it then took 10-15 years to design, develop, and test the drugs we use today in standard-of-care treatment.
Once a target is identified, the next step in the project is to design and then synthesize a molecule that one hopes will exhibit all the required properties of a good drug; such a molecule is referred to as a drug candidate. Drug discovery is incredibly difficult because the candidate has to pass many tests before clinical trials (where the drug is in most cases tested first in healthy volunteers and then in patients) are even allowed to start – and then has to succeed in the clinic. Firstly, it has to inhibit (or activate) the target, strongly enough to have the desired biological effect. But it cannot interfere with the function of any other proteins nearly as strongly, because that is one of the main causes of undesirable side effects. An oral drug (a drug taken by mouth) must diffuse from the stomach into the organs of the body where the inhibition is required, without getting digested along the way. Once arriving at the cell, the drug must cross the cell membrane (which is composed of lipids), an often difficult task. Finally, the drug has to be metabolized and excreted on just the right timescale; not too quickly (or the biological effect would be eliminated), but not too slowly either (or its effects might linger far past the desired treatment period).
Many current drugs are far from ideal, because they produce various types of side effects, some of which are quite unpleasant and on occasion fatal. The potential high impact use of computer models in drug discovery can now be readily inferred; what if the key properties of the candidate drug molecules could be calculated, at a cost far lower per compound than the $5000 required for experimental synthesis and testing? It has been estimated that there are 1060 possible drug candidates that could be synthesized. (To get a sense of how big this number is, there are 1021 stars in the universe and there are 1046 water molecules in the earth’s oceans.) Somewhere in that enormous list of molecules is almost certainly one with better properties (likely much better properties) than those created using purely experimental approaches. The challenge is to predict, using computers and accurate scientific algorithms, the key properties a drug candidate must have.
In applying computational methods to drug discovery, the initial goal would be to predict potency, i.e., how tightly a proposed drug candidate binds to the specified protein target; very tight binding is necessary in order to inhibit (or activate) the target at low enough doses of the drug such that the function of other proteins in the body are not impacted. An exciting possibility would be to build a physical model of the drug molecule interacting with the protein target at an atomic level of detail, and then calculate the strength of the binding using the laws of physics. Physics-based simulations at the nanoscale – that is, simulations that incorporate an atomic level of detail of the physical system – have been used in pharmaceutical research for more than 30 years, but as a methodology for making quantitative prediction, the results were for many years disappointing. Until quite recently, the calculations lacked the accuracy and robustness needed to elucidate such differences, and thus engineer superior molecules.
Ten years ago, Schrödinger, Inc., set out to develop an accurate and reliable physics-based solution to the problem of drug binding simulation, building on years of earlier work in academic laboratories. Over the past several years, we have seen an inflection point in the technology; accuracy is now consistently high enough to effectively be used to guide real-world drug discovery projects. It is now possible to predict the binding affinity of a drug candidate, using a “computational assay”, for as little as $5 per molecule, as compared to the $5000 required to make and test the compound experimentally. Furthermore, this cost can be expected to diminish with time, following Moore’s law.
Physics-based simulation methods are already widely used in important engineering applications. No one would build a bridge or tunnel without appropriate calculations of the stresses and strains on the proposed structure. Computer simulations of airflow have replaced wind tunnel experiments in the design of airplanes, such as the Boeing 787 Dreamliner. The flow of electrons in a microprocessor is modeled by sophisticated simulation packages that provide approximate solutions to the Schrödinger equation for crystalline semiconductors.
The challenge of simulating a drug molecule binding to a protein target is considerably more difficult than any of the above problems. Of the three problems described above, only the microprocessor simulations involve nanoscale modeling, and this problem is simplified by the regular structure, and rigidity, of the silicon crystal lattice. In a drug binding simulation, hundreds of thousands of atoms have to move in a complex, irregular dance that must faithfully reproduce the microscopic interactions prescribed by the relevant laws of physics (e.g., the Schrödinger equation).
Physics based simulation is the bridge between the world of bits, in which Moore’s law holds sway, to the world of atoms and molecules, which has for the most part stubbornly resisted the kind of exponential improvement that gave us the Internet and the iPhone. It is the key to digitally enabled drug discovery and, ultimately, all sorts of new materials and processes.
When Schrödinger, Inc, was founded in 1990, the initial goals were modest: commercialize a new quantum chemistry program developed in Prof. Rich Friesner’s academic laboratory at Columbia University. Funded primarily by NIH Small Business Innovative Research Grants, this effort was a success; by 1998, the company had annual sales of approximately $1 million from selling quantum chemistry tools that were 3-5 times faster than the current state of the art to pharmaceutical and biotechnology companies.
In 1998, classical (vs. quantum) mechanics technology (often referred to as molecular mechanics) was acquired from another Columbia University group, and David Shaw, a computer scientist who founded the D. E. Shaw group and currently serves as Chief Scientist of D. E. Shaw Research, made a significant investment in the company. A decision was made to focus on the development of the next generation of tools for drug discovery, and to create integrated solutions capable of quantitative accuracy. Even at the time, it was clear that the latter task was a long term project that would require patient capital, given the need for better functions for describing interatomic energy potentials, faster and more robust simulation algorithms, and considerably more computing power. Fortunately, Schrödinger had a strategic investor who believed that ultimately, digital simulation had the potential to revolutionize the drug discovery enterprise as it had many other industries. A number of years later, in 2010, Bill Gates became a major investor in Schrödinger; he shared a similar long term vision and commitment to the transformative nature of the scientific and technical opportunities. Schrödinger could not have successfully embarked on, or completed, the journey described here without the level of confidence, and financial backing, provided by Shaw and Gates.
By 2008, Schrödinger had become a major player in the molecular modeling space, with a number of best-in-class software solutions that were widely used by pharmaceutical modeling groups in drug discovery projects. However, it became apparent that to make further progress, Schrödinger was going to have to work on drug discovery projects itself.
In 2010, Schrödinger and Atlas Ventures, a leading life science venture capital firm, formed a joint venture to push the boundaries of computationally driven drug discovery. The resulting company, Nimbus Therapeutics, has provided an initial picture of what state of the art computation is capable of. Of the three Nimbus projects that have been operative for more than a year, one was partnered with Genentech; the second was sold to Gilead Sciences for $1.2 billion and yielded a compound that recently entered Phase 2 clinical trials; the third has effectively addressed a seemingly intractable problem in compound design, and is advancing toward the clinic.
When Nimbus was started in 2010, the average computational chemistry effort per drug discovery project in large pharmaceutical companies was on the order of 0.25 FTEs. This modest effort reflects the auxiliary role played by computation on project teams. Projects run in Nimbus have employed a significantly higher level of computational effort, as well as having accessibility to many more software licenses of key tools and a much higher level of processing power. Of equal importance, the operational protocols in a drug discovery project have been optimized to exploit the rapidly increasing capabilities of modeling methodology. An iterative feedback loop was established in which experience in projects drives improvements in the software, which in turn engenders further optimization of protocols.
The use of computation in advancing projects can be divided into four types of applications:
(1) Target analysis: Schrödinger has developed specialized protocols to assess the druggability of a protein target; these have been essential in improving the probability of success.
(2) Virtual screening: Schrödinger’s virtual screening tools are widely acknowledged as the best in the industry. Virtual screening involves scanning a large collection of purchasable compounds (currently about 10 million) using fast approximate methods to identify initial hits that can then be optimized into a drug molecule in subsequent stages.
(3) Candidate design: Once initial hits have been obtained, they have to be chemically modified to achieve all the required properties of a drug, including much tighter binding affinity to the protein target. In a Nimbus project, this process entails investigating hundreds of thousands of “design ideas” using a range of computational tools, and involves an intimate collaboration between Nimbus synthetic chemists and biologists, and the Schrödinger computational modelers. These collaborative interactions have been facilitated by a new enterprise software application developed by Schrödinger that enables design ideas to be exchanged, annotated, stored, and viewed by the entire team. Only candidates passing computational filters in the design process make it to the next stage.
(4) Computational assays: When the precision of the calculation of an important quantity, such as the binding affinity of a drug candidate to the protein target, reaches a level that is comparable to the estimated error in experimental measurements, the calculation moves from the status of a tool (where the errors are larger in magnitude and more unpredictable) to that of an “assay” – a filter that can be relied on from a statistical point of view to reliably evaluate the property in question. Schrödinger has promoted an increasing number of computational protocols to assay status over the last several years. The emergence of accurate computational assays is a novel development that is a key catalyst for dramatically increased value creation in computationally driven drug discovery projects.
Over the past 6 years, Nimbus and Schrödinger have integrated these components into a highly effective platform that continues to evolve in its ability to drive projects forward.
Acetyl-CoA carboxylase (ACC) is a critical enzyme involved in lipid metabolism. In animal models, blocking ACC forces cells to metabolize fat, thus leading to weight loss and reduction of fat in the liver. Blocking ACC also selectively kills specific types of cancer cells that are dependent upon an ACC-based metabolic pathway for survival. There are thus many disease indications that could in principle benefit from treatment with a molecule that inhibits ACC. Nimbus has chosen initially to focus on nonalcoholic steatohepatitis (NASH), which currently has no effective treatment, and can if untreated ultimately lead to more serious liver disease (cirrhosis, liver cancer). NASH at present affects 2-5% of Americans.
The Nimbus/Schrödinger collaboration succeeded in developing a highly promising ACC inhibitor, despite the fact that other companies have failed to make progress against this very difficult target. The first key decision was to target the inhibitor at an unusual allosteric binding site of the protein (i.e., a site that is not where a substrate of the enzyme would bind), as opposed to the normal site of enzyme activity. Schrödinger target analysis indicated that the normal site was undruggable, whereas the allosteric site was amenable to the right type of molecule. A virtual screen then located a viable lead compound, which was transformed via the design/optimization process into a development candidate in just 18 months, after synthesizing only a few hundred compounds (a typical project requires synthesis of several thousand compounds en route to a development candidate, and takes 3-5 years or longer). The compound successfully passed GLP toxicity testing in 2014, entered Phase 1 clinical trials in 2015, and entered Phase 2 clinical trials in late 2016.
The combination of safety profile, initial evidence of efficacy, first in class inhibition against a novel target, and potential to address a large, unmet medical need, engendered substantial interest in the compound from major pharmaceutical and biotechnology companies. Ultimately, a deal was concluded with Gilead Sciences, currently the leader in treating liver disease with its Harvoni medication for hepatitis C. The purchase price was $400M upfront and $800M contingent upon future successful clinical outcomes. A $200M milestone payment was recently made when the clinical compound entered Phase 2. Further discussion of the ACC project, including economics for the investors, can be found in this blog written by Bruce Booth, the Atlas partner who co-founded Nimbus with Schrödinger:
Schrödinger is now engaged not only in follow on projects with Nimbus, but with a wide range of pharmaceutical, biotechnology, and venture capital partners. The success of the ACC program, coupled with a suite of accurate and robust computational assays validated over the past several years (see below), has generated enormous excitement in the industry.
In the early 1980’s, a specialized type of simulation, called free energy perturbation (FEP) was developed to improve the efficiency of rigorous binding affinity calculations. An FEP calculation starts with a reference molecule – a drug candidate whose binding affinity is already known from experiment. Since a drug discovery project is always about improving an existing candidate molecule, a suitable reference is available. During the simulation, the reference molecule is modified, by an “alchemical transformation”, into the new molecule of interest. In addition to binding affinity, FEP can also be used to assess other important properties, such as the solubility of a molecule in water.
By 2010, the conditions for developing an accurate and robust FEP methodology that would be useful across many drug discovery projects were in place. Computing power had reached the point where efficient physics-based simulations, using classical mechanics, could be performed, and more than 50,000 protein structures were available in the public repository of proteins structures, the Protein Data Bank (PDB), including a large number of interesting drug targets. Schrödinger identified the following key systematic improvements required to bring FEP to computational assay status, and launched a large-scale engineering project to address each of them:
(1) development of the first truly comprehensive and accurate molecular mechanics models covering medicinal chemistry space – such models are essential in representing proteins and drug molecules in a classical computer simulation;
(2) specialized techniques to make sure that during the course of the simulation, the system is able to visit all important drug candidate/protein receptor structures without requiring excessive computation time;
(3) development of a graphical user interface (GUI) enabling ease of use; prior to development of the GUI, it would take weeks to months to properly prepare a single calculation; the Schrödinger FEP user interface enables hundreds of calculations to be launched in a few hours;
(4) tools for accurate initial setup of the system, including placement of key water molecules and hydrogen atoms, and the initial positioning of the new drug candidate molecule; and
(5) efficient performance of the simulation code using graphical processing units (GPUs); for physics-based classical simulations, GPUs provide a factor of roughly 10x improvement in cost/performance as compared to traditional CPU-based architectures.
It has been demonstrated through large-scale retrospective and prospective testing on more than 100 protein targets and ~5,000 molecules that our implementation of FEP provides a computational assay that is nearly equivalent to performing the corresponding experimental assay. In approximately 50% of cases, the prediction is within the typical error of experimental measurements; in 90% of cases, the prediction is within a factor of 10 of the experimental measurement. This level of accuracy is high enough to have a very significant impact on driving drug discovery projects. There are now a very large number of examples from our collaborators where FEP was successfully used to advance a program that might otherwise have taken a much longer time to progress to the same point or even failed to produce a viable drug candidate.
The impact of FEP can be dramatically increased by scaling up the number of calculations. One situation where this is an essential strategy is when the target is a very difficult one on which to make progress. A recent project in this category is an ongoing collaboration with a top 5 pharmaceutical company. The collaborators had been unable to make any progress in improving potency, starting from an interesting lead compound derived from experimental screening, despite having synthesized 73 compounds.
3500 compounds were run through the FEP computational assay. Experimentally, it would have taken a minimum of several years, and on the order of $10M, to make and experimentally assay the same number of compounds. The cost of the FEP calculations was approximately $20,000. Of the 3500 compounds, only 23 were predicted to improve the binding affinity of the lead compound. This is consistent with the difficulties experienced in a traditional experimental approach to the problem. To date, the top three compounds selected by FEP have been synthesized and tested experimentally. Two of the compounds have provided more than an order of magnitude increase in potency, while results for the third are still being analyzed.
The results described above demonstrate that drug discovery projects in which the structure of the protein target is known are now enabled on the Moore’s law curve. This means that in 10 years, the cost of FEP calculations is expected to fall by approximately a factor of 30 (assuming a 2x reduction in cost/performance every two years). Alongside the hardware improvements, we can expect both speed and accuracy increases from improvements in the software and algorithms. More properties will be amenable to a computational assay, and the existing assays will converge to uncertainties equivalent to those inherent in experimental assays. Fast approximate methods will also improve, enabling the preliminary screening of billions (or even trillions) of compounds to become more robust. All of this implies that the quality of development candidates (and hence the success rate of new drug candidates in the clinic) will increase, while project costs will at the same time decrease. The counterweight of highly challenging biology and a higher safety/efficacy bar for new drugs will still be present; but reversing Eroom’s law using accurate, robust nanoscale simulations appears to be likely.
Not every project is currently amenable to this technology due to lack of an available protein structure, but an increasing number are as more protein structures are determined, and experimental techniques improve. Exciting new methods based on cryogenic electron microscopy (cryo-EM) are facilitating the determination of larger, more complex structures on an increasingly routine basis. This means that the number of projects that can benefit from atomistic simulation will grow every year.
The discussion above has focused on small-molecule drug discovery efforts, but the same techniques can be applied to optimization of more complex pharmaceutical agents, including macrocycles, antibodies, and other protein therapeutics. Encouraging preliminary data along these lines has already been produced.
More generally, personalized, or precision, medicine requires the design of molecules customized to the patient’s genetic profile; this in turn implies development of a much larger number of molecules, with fewer patients served by each molecule. A combination of high precision drugs, along with drastically lower development costs, will be needed to make this vision a reality, as a complement to “big data” genomics analysis. Nanoscale biomolecular simulation can deliver on both of these requirements, opening up large new market opportunities.
Schrödinger’s goal is to create an encompassing systematic approach exploiting the revolutionary implications of digitally enabled drug discovery. Such an approach involves deep integration of experimental methodology with computational assays, a wide range of auxiliary modeling technologies, and a collaborative enterprise platform for managing experimental and computational data, as well as enabling rapid digital prototyping and testing of new designs prior to expensive experimental synthesis and testing. The success of the Nimbus ACC project, followed by the development of a widely effective FEP computational assay, represents the leading edge of a strategic inflection point, which Schrödinger is well poised to capitalize on going forward.
Nanoscale simulation can be applied to many materials and process design problems other than drug discovery. At present, these applications are somewhat behind drug discovery; however, success in drug discovery will accelerate the adoption of the technology in fields as diverse as electronic materials, polymers, catalytic chemistry, and energy conversion.
In a recent book, economist Robert Gordon makes the case that GDP growth over the past 50 years has dramatically slowed as compared to the previous 100 years due to lack of productivity growth in major segments of the economy. While we are all dazzled by the changes in information technology, the world of bits is quantitatively limited in size as compared to the world of atoms. In the end, the limiting factor in electric cars revolutionizing transportation is the capabilities of the battery, not self-driving software. And battery improvement has proceeded slowly over the years, despite massive capital investment.
Is there a combination of materials that would yield a battery that is 2x, 3x, or even 10x more capable and cost effective than the lithium ion units currently in use? No one knows for sure, but the fraction of materials that have been screened experimentally is miniscule compared to what is theoretically possible. A computational assay for battery performance is not feasible at present; but things can change very rapidly when dealing with exponential growth. When the world of atoms as a whole is enabled by Moore’s law, a new industrial revolution may once again reset the human ability to transform nature for the better.
The strategic inflection point in nanoscale simulation presents investors with an opportunity to ride a wave of rapid innovation and exceptional value creation. Computationally driven projects can put superior molecules in the clinic at a fraction of the cost of an equivalent experimental effort. For this reason, Schrödinger is experiencing exponentially growing interest in joint ventures at increasingly attractive deal terms. Additional capital will enable Schrödinger to scale up its portfolio of risk sharing projects, in both number and in the magnitude of the equity stakes. Risk sharing requires up front capital investment, but delivers highly leveraged rewards for success in the clinic. Projections of current opportunities indicate that a revenue stream in excess of $200M/year from drug discovery partnerships could be reached within 4 years, an amount that could be significantly increased with access to additional investment capital.
Software sales benefit from the success of risk sharing ventures by illustrating the transformative nature of the strategic inflection point, and by changing the market power equation in large customer relationships. We expect software sales to grow at a minimum of 10-15% per year, and quite possibly at a significantly higher rate as FEP+ deployment is scaled up, and our enterprise platform gains traction. New investment capital will enable the relatively modest increase in sales, support, and infrastructure required to grow the software business. The steadily-increasing renewable revenue stream from this business is complementary to the more rapidly growing, but less predictable, risk sharing revenue stream.
Finally, continued investment in cutting edge science and technology development, scientific software engineering, and enterprise platform functionality are essential in order for Schrödinger to retain a leadership position in nanoscale simulation and modeling. The major fraction of returns is generally captured by the market leader during a strategic inflection point. Our research pipeline indicates that breakthroughs similar to FEP are possible in the next 5 years across a wide range of compelling applications, including protein structure prediction, development of computational assays for antibodies and other biologics, application of deep learning to drug discovery, and design of new materials. Schrödinger’s demonstrated ability to translate innovative basic research advances into robust, high performance software will ensure the continued success of drug discovery partnerships, loyalty and increased subscription of software customers, and penetration into new markets.
Richard Friesner, Ph.D

Together with Sanofi, Schrödinger recently published a manuscript in the Journal of Medicinal Chemistry, titled “Automated Design of Macrocycles for Therapeutic Applications: From Small Molecules to Peptides and Proteins.” The paper presents the implementation and application of a new in silico workflow for the efficient and automated design of novel and diverse macrocycles.
We sat down with Drs. Andreas Evers (who has since moved to Merck KGaA / EMD Serono) and Michael Wagner of Sanofi, as well as Dr. Dan Sindhikara of Schrödinger to discuss the paper.
Tell us what’s new and key in this paper?
Andreas: In this paper we describe a brand-new workflow to automatically generate, evaluate, and propose synthetic strategies to create macrocycles with desired properties. As you know, macrocycles and cyclic peptides are increasingly attractive therapeutic modalities, and a number of macrocycles with a molecular weight ranging from 500 to 1500 Daltons have become successful drugs, with several examples that can be administered orally. We see macrocyclic molecules as filling an important gap in the world of drugs between small molecules and larger biologics.
As described in our paper in detail, our procedure proved to be very effective, and can be applied for the cyclization of small molecules and peptides and even PROTACs and proteins. We are very excited by the promise shown in our applications of the technology so far.
That’s very interesting, can you tell us how you came to work with Schrödinger on this?
Michael: Within our group at Sanofi, Synthetic Medicinal Modalities, we were interested in coming up with a way to rationally design macrocycles, and we wondered if we could create a method to suggest synthetic strategies for cyclization that would take advantage of well‑established chemical reactions and reagents.
Andreas: At Sanofi, we’ve used Schrödinger’s computational platform for many years on various projects, and while there were tools for studying macrocycles, there was nothing for efficient screening of different attachment sites and possible linkers for a linear starting molecule to automatically design new macrocycles. That’s when we reached out to Dan, and discussed with him what we had in mind, and Dan helped to create this automated workflow, and throughout the project, we worked closely together to validate the new method, as well as to suggest improvements to the technology.
Can you tell us a little more about the workflow?
Dan: Sure, you can find the full details of our integrated approach in our paper, but generally, it involves several steps as this figure illustrates (see below).
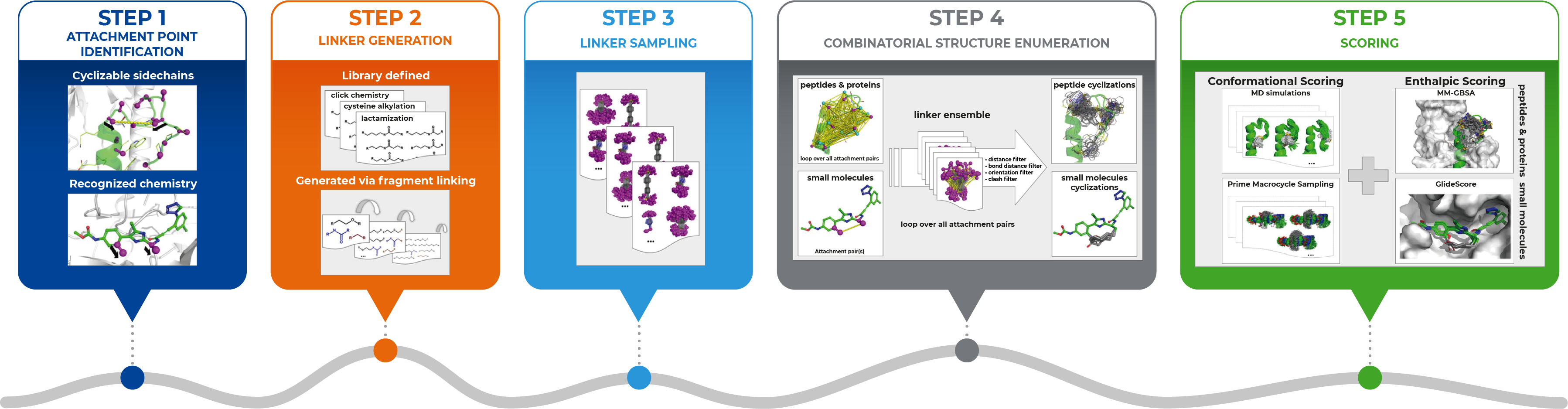
We begin in Step 1, where we use the bioactive conformation of a linear molecule as the starting point – the attachment sites may either be identified automatically or specified by the user. Then in Step 2, we generate the chemical linkers – these may either be provided as explicit lists, e.g., based on available chemical reagents, or optionally, the workflow could generate them automatically by combining smaller fragments. In Step 3, we generate the full conformational ensemble of every linker, and in Step 4, we map the ensemble of linkers to the attachment points, and eliminate unfavorable cyclizations that are not geometrically feasible. Finally, the cyclized ligands can then be ranked through conformational and/or enthalpic scoring. The approach involved using features in various Schrödinger Software including Prime, Desmond, and Glide.
I should also point out that it is possible to generate bicyclic structures in a stepwise manner with our approach, as seen in the retrospective study of the KIX Domain from the Human CREB Binding Protein. We investigated and found that our in silico cyclization approach was able to automatically generate the known bicyclic construct. The case also demonstrates our method’s ability for the engineering of proteins for increased tolerance toward thermal and chemical
denaturation.
Andreas: We put particular emphasis on assuring the synthetic feasibility of the resulting proposals. For this purpose, we compiled an explicit list of linkers based on commercially available chemical reagents. But the list can also be easily modified by the user to consider specific cyclization chemistries and reagents. In addition, we also built in another option that instead of specifying predefined cyclization linkers, the linkers can be constructed in silico from a small set of reactive “breeding fragments,” that can be combined and enumerated to fit within the geometrical constraints of the ligand-receptor complex. As a result, the algorithm will suggest a diverse set of linker chains that are synthetically tractable.
How well did the method perform? What do you see are the key advantages?
Andreas: We carried out several prospective and retrospective studies to validate our approach against different targets and modalities of pharmaceutical interest, and found excellent agreements with experiment. And the workflow automated much of the tedious manual work we used to have to do, and allowed us to more broadly and systematically explore the chemical space for possible attachment sites and linkers.
Dan: Our approach has “synthetic intelligence” built in so that it not only produces novel molecules, but also the list of chemical reagents for their synthesis. In collaboration with the synthetic chemist, the user has the option to reduce or extend the list of reagents, for example, by focusing on a specific cyclization chemistry or considering only reagents that are physically available or can be quickly procured. By doing so, we ensure the resulting design proposals can be straightforwardly synthesized.
Do you think you will use these tools routinely in your drug discovery efforts?
Michael: Definitely, in fact, as we showed in the paper, we used the technology prospectively in two projects – a peptide (dual GLP-1/glucagon receptor agonists) and a small molecule example (TAFIa inhibitors). And in both cases, we were able to design molecules that were novel and highly potent. And the fact that the design proposals can be straightforwardly synthesized is highly appreciated by the medicinal chemists. Therefore, the approach as such is meanwhile used in a standard way in all drug discovery programs where molecules could benefit from cyclization, either in terms of potency, intrinsic stability, or other properties.
Given that until now we’ve never had such a workflow that can propose easily-synthesizable cyclic molecules in an efficient manner before, I believe we can significantly accelerate the cycle times within a drug discovery program.
Besides the demonstrated success in proposing easily-synthesizable macrocycles with desired properties, is there anything else that you find exciting about the potential of this new technology?
Michael: Based on the success in our retrospective studies on systems larger than small molecules and peptides, we believe this approach holds great promise in being applied to other modalities such as PROTACs or even proteins, for example, to improve their thermostability, which is often an important goal in engineering studies for proteins/enzymes for biotherapeutic or biotechnological applications. Currently, we are applying this approach on several internal projects with great initial success.

Principal Scientist Computational and Structural Bioinformatics, Merck KGaA

Director Medicinal Chemistry, IDD Synthetic Medicinal Modalities, Sanofi-Aventis

Principal Scientist, Technical Lead Life Science Software, Schrödinger
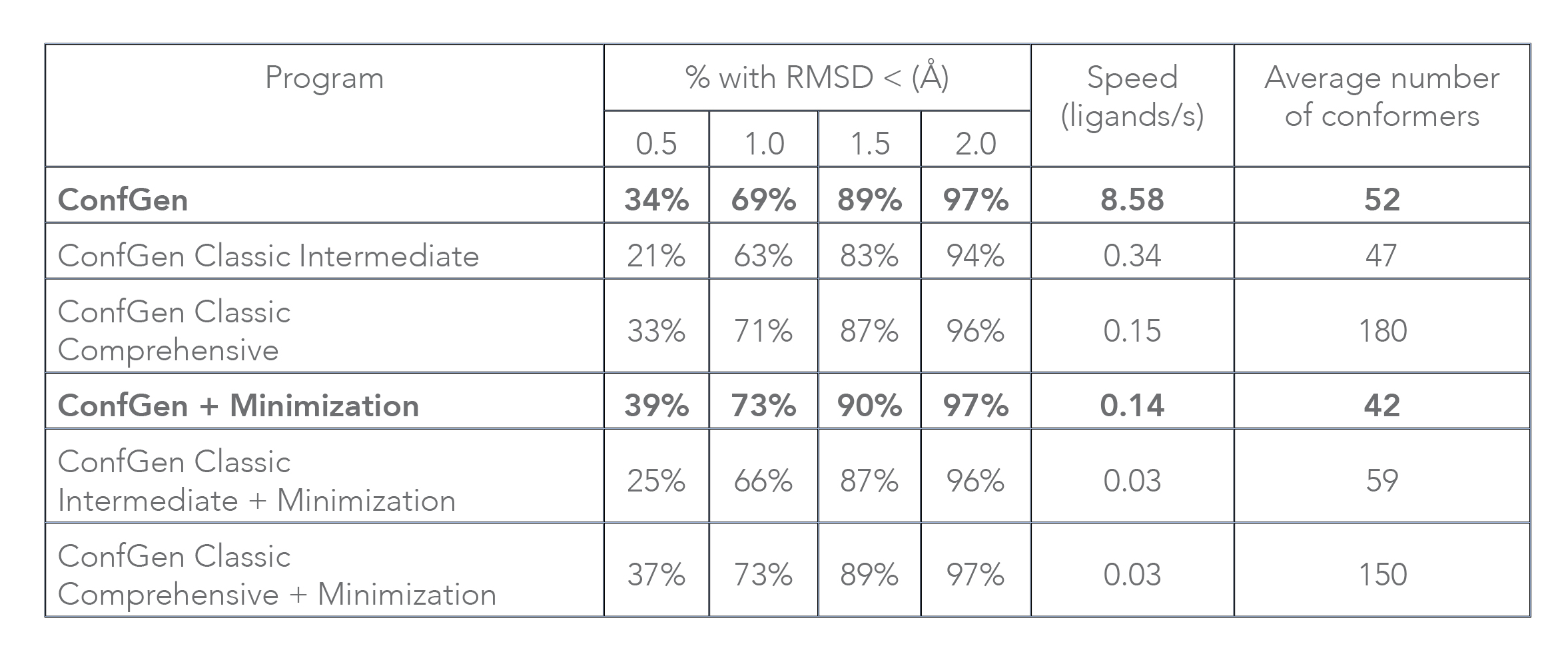
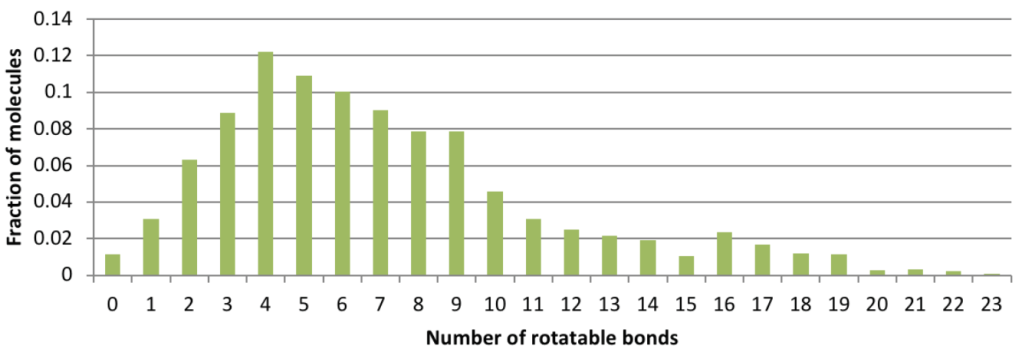
Conformation generation, a procedure for producing diverse, low-energy 3D structures of compounds, is an essential element of in silico drug design, heavily employed in ligand-based virtual screening. To improve the recovery of bioactive conformations among generated conformers and drastically speed conformer generation, Schrödinger has completely rewritten the ConfGen application. As shown in Table 1, the new algorithm is approximately one order of magnitude faster than the previous version of ConfGen (henceforth referred to as ConfGen Classic), with bioactive recovery equivalent to the most computationally intensive mode of ConfGen previously available. This case study utilized 1,904 ligands, extracted from high quality protein-ligand crystal structures. This dataset includes a broad diversity of functional groups present in drug-like molecules and a range of molecule sizes and flexibility, as shown in Figure 1.
Without minimization of output conformers, ConfGen finds the bioactive conformation with an RMSD of < 1.5 Å in 89% of cases compared to 83% and 87% with ConfGen Classic Intermediate and Comprehensive modes, respectively. The new algorithm processes ligands 25 and 57 times faster than ConfGen Classic Intermediate and Comprehensive modes. When minimization of conformers is performed, ConfGen recovers more bioactive conformations with < 1.0 Å RMSD than without minimization. Because minimization acts as the speed bottleneck, the new algorithm is 5 times faster than both ConfGen Classic Intermediate and Comprehensive modes. It should be emphasized that ConfGen is able to achieve bioactive recovery similar to ConfGen Classic Comprehensive in far fewer conformers.
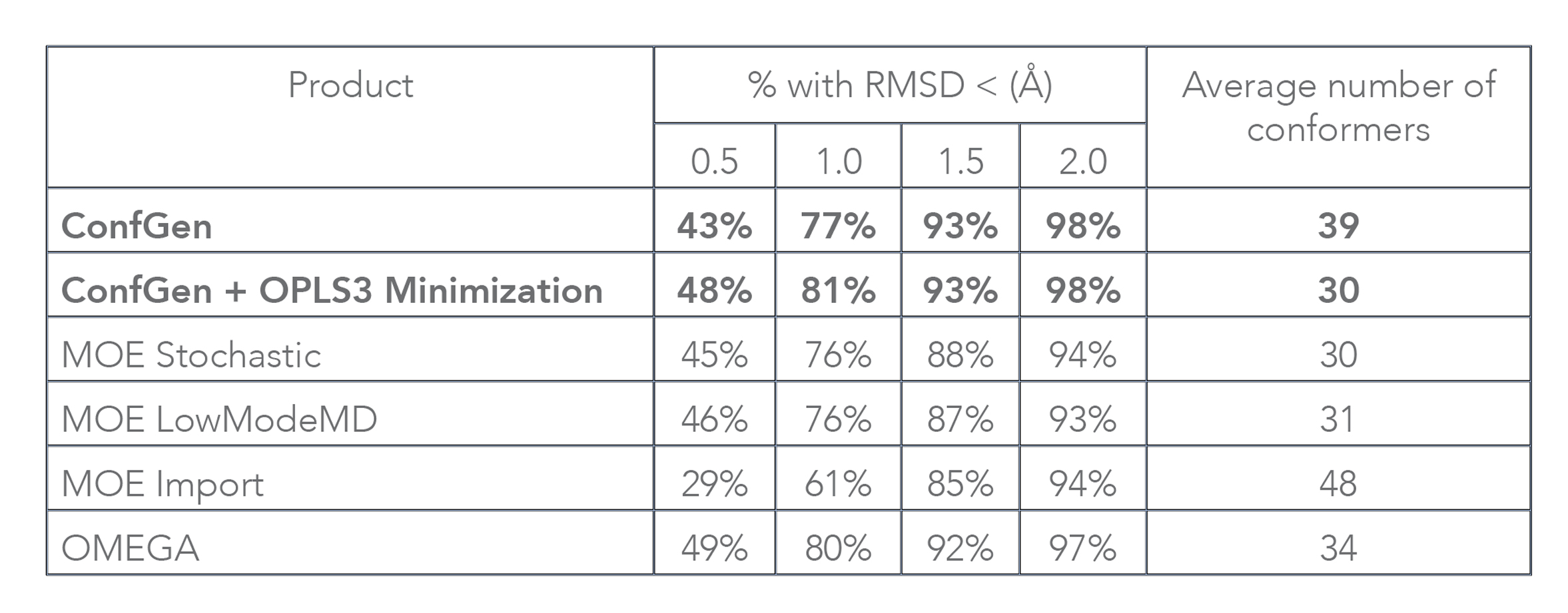
Both ConfGen and ConfGen Classic, along with several other commercial conformer generators, have been benchmarked at the Universität Hamburg [1] on a high quality dataset of 2,859 protein-bound ligand conformations extracted from the PDB. In terms of accuracy, the ConfGen program was found to be on par with OMEGA and overall better than MOE (see Table 2). Note that this paper assessed processing time by running conformer generator programs for a single ligand at a time, which is not representative of typical use. In addition, overhead from Schrödinger’s job-control system that enables easy submission to remote queues and hosts was included as part of the ConfGen timing. With these contributions factored out, when applied to the dataset used in that study, ConfGen (Suite 2017-3 release) processes on average 15 ligands per second without optimization, and about 0.4 ligands per second with geometry optimization using OPLS3. These speeds are similar to OMEGA’s and at least an order of magnitude faster than MOE Import, the fastest MOE approach.
The new ConfGen algorithm utilizes a divide-and-conquer strategy to build feasible molecular conformations in three steps: (1) divide input molecules into fragments by breaking exo-cyclic rotatable bonds; (2) obtain conformations for all the fragments; (3) build conformations for the whole molecule by reconnecting the fragments in various ways.
The following types of bonds are considered non-breakable for fragmentation purposes: ring bonds, double or triple bonds, bonds to terminal atoms, and common types of “rigid” bonds such as N-C bonds in amides and thioamides and O-C bonds in esters and thioesters. All other bonds are broken and capped by hydrogens during fragmentation. This process is illustrated in Figure 2.

An examination of the Zinc database [2] was used to identify approximately 40,000 template fragments. A MacroModel conformational search was applied to each of these fragments to generate a library of conformations for use in ConfGen searches. Conformations for fragments not found in the library are generated on the fly via a two-stage optimization process starting from random initial atom positions.
The first optimization stage uses a penalty function to efficiently obtain crude coordinates while the second stage uses the OPLS3 force field to fine-tune the conformations. The process is repeated a number of times depending on the number of sp3 centers in the fragment. By studying compounds from several public databases, it was observed that at most roughly 2n⁄2 low-energy conformations, where n is the number of sp3 centers, are expected for the fragments generated using the ConfGen fragmentation rules. The algorithm generates this many conformations up to a maximum of 24 by default. Conformations with heavy atom RMSD values less than a configurable threshold (0.25 Å by default) relative to previously generated conformations are classified as duplicates and discarded.
The penalty function used in the first step of the on-the-fly conformer generation for fragments consists of terms designed to produce the desired features of the conformations:
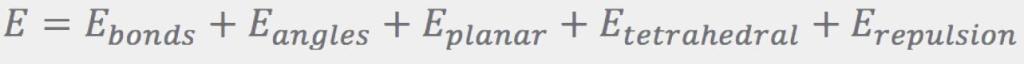
The Ebonds contribution is a sum of harmonic terms (one for every bond) that penalize departures of the bond lengths from their ideal values:
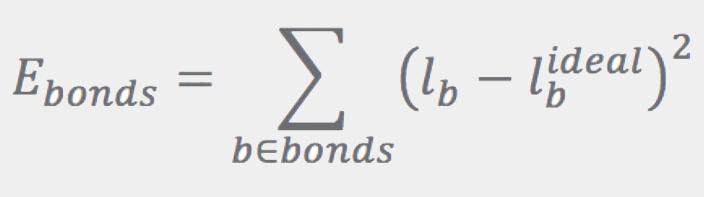
Ideal bond lengths, lbideal, are obtained as described in Refs. [3] and [4]. The Eangles term has the same form as Ebonds, except that the sum runs over angles. Ideal angles are determined using simple rules based on atom hybridization, number of connections, and ring membership. Instead of using the angles directly, Eangles uses the distance between the first and third atoms in the angle, la:
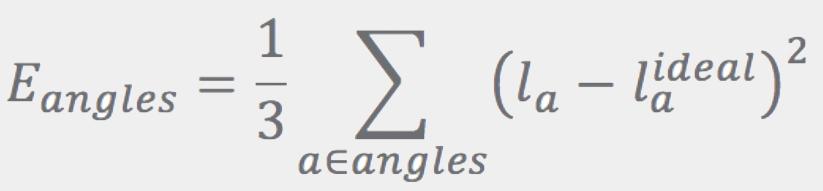
Both of the Eplanar and Etetrahedral terms are given by the sums over “templates”:
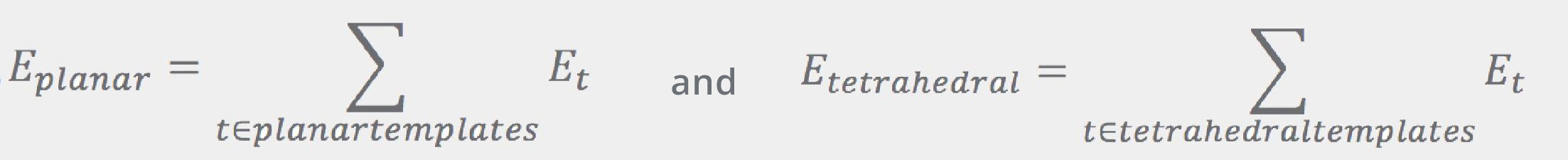
where Et is proportional to the RMSD of the fragment’s atoms with regard to the corresponding atoms in the template:
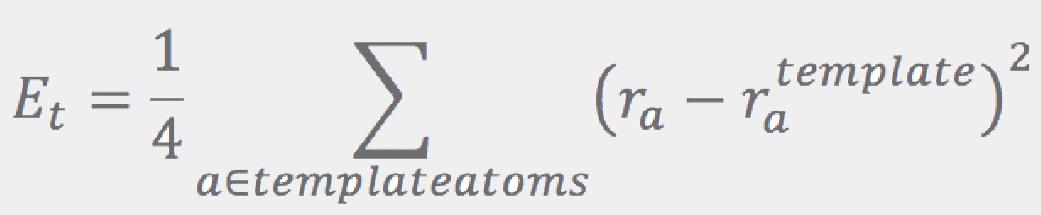
Here, template atoms are assumed to be superimposed onto the corresponding fragment’s atoms. ConfGen uses three kinds of templates: one for tetrahedral centers (3 or 4 atoms around an sp3 center) and another two for the planar configurations around sp2 atoms and double bonds. In all cases the ideal (template) positions are built using ideal bond lengths and “perfect” angles (120° for the planar templates and 109.5° for the tetrahedral). Finally, the Erepulsion term is given by a sum of short-ranged soft “bumps” over the non-bonded atom pairs (that is, pairs of atoms that do not contribute to Ebonds or Eangles):
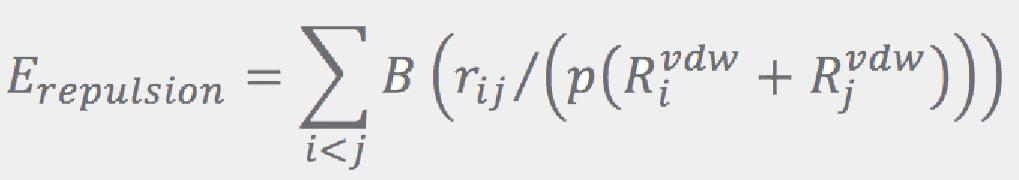
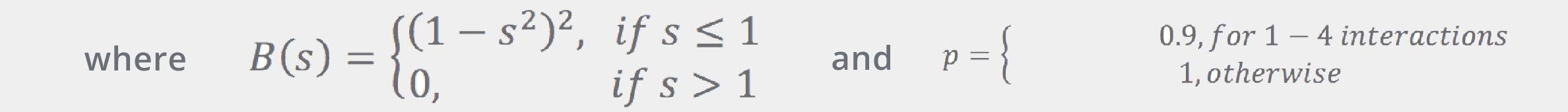
where rij are the interatomic distances, and RivdW are the van der Waals radii (from Ref.[5]).
The fragments are reconnected along bonds broken in the fragmentation step, one bond at a time. The linking order is chosen to favor joining similarly sized portions of the molecule at each intermediate step. Specifically, at each level, conformations for two fragments (A and B) are used to create the conformations for the combined fragment A+B. First, SMARTS patterns matched against the whole molecule are used to select specific dihedral angles to sample along the A − B bond. When there is only one conformation available for either (or both) of the fragments, the fragment’s symmetry is first identified by rotating it along the new bond by 45, 60, 90, and 180 degrees to see if equivalent structures result.
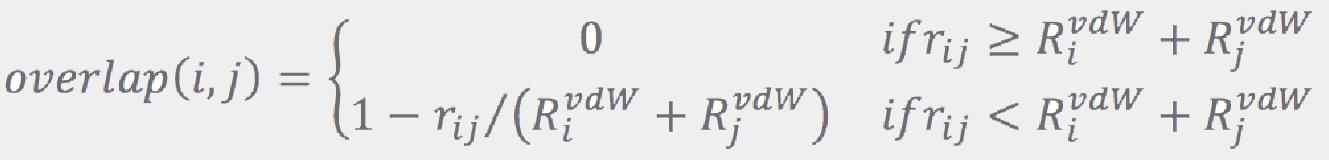
where rij is the distance between atoms i and j, and RijvdW are their van der Walls radii from Ref. [5]. If the largest overlap is less than a configurable threshold (0.5 Å by default), the combination is accepted as a viable candidate. This process is repeated until a prescribed number of candidates (96 by default) is collected. If no combination passes the overlap test, all of them are considered to be viable candidates. A prescribed number of the best ranked candidates is then retained (64 by default), while the others are dropped. More conformations are retained for larger molecules: if the number of rotatable bonds exceeds some threshold (9 by default), the values regulating number of conformations (number of conformations for a fragment, number of candidates, and number of conformations to keep after each “join”) are increased by a factor that is a function of the number of the rotatable bonds.
Conformations are ranked using a sum of Lennard-Jones 6-12 non-bonded potentials (for rapid processing the L-J parameters are obtained from a look up table of DREIDING force-field [6] values), ad-hoc tabulated dihedral penalties associated with the inter-fragment bonds, and contributions that facilitate equatorial (versus axial) ring attachment configurations by enhancing specific 1-4 interactions. The process of joining progressively larger fragments together and sampling multiple orientations for the restored bond continues until the entire molecule is reconstructed. Duplicate conformer elimination is only performed for molecules smaller than a certain threshold (having less than 32 heavy or polar hydrogen atoms by default) because this process is slow and rarely impactful for the large molecules.
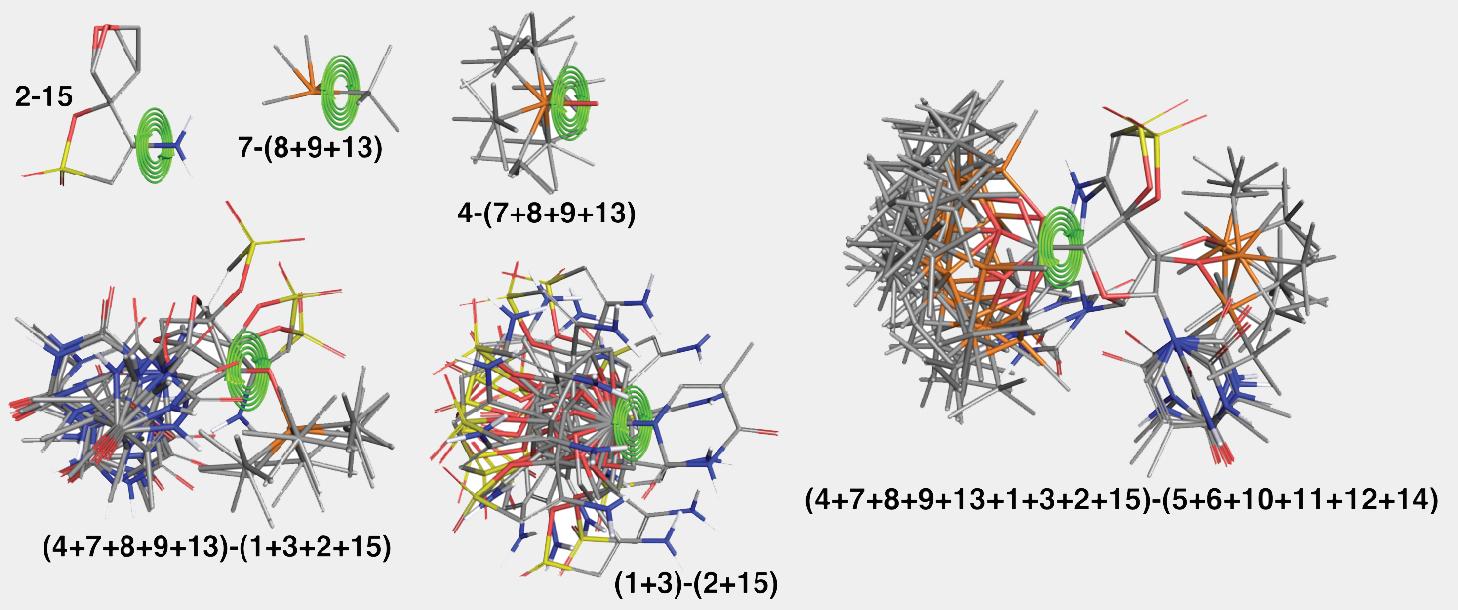
Key steps of the algorithm are illustrated in Figure 2 and Figure 3 for the ligand from PDB structure with code 3QO9. The structure is split into fifteen fragments as shown in Figure 2. Only the fragment labeled as “2” has more than one low-energy conformation. The algorithm picks the order in which fragments are assembled to minimize the number of the atom pairs that would need to be checked for overlaps. For this structure the order was as follows: 1+3, 2+15, 5+6, 9+13, 12+14, 8+(9+13), 11+(12+14), (1+3)+(2+15), 7+(8+9+13), 10+(11+12+14), 4+(7+8+9+13), (5+6)+(10+11+12+14), (4+7+8+9+13)+(1+3+2+15), (4+7+8+9+13+1+3+2+15)+(5+6+10+11+12+14). Many assembly steps involving simple fragments, e.g., 1+3, 5+6, 12+14, etc., resulting in a merged fragment that has only a single distinct conformation. Illustrative stages of the assembly algorithm are shown in the Figure 3.
Several ConfGen parameters can be used to control tradeoffs in calculation speed and recovery of bioactive conformations. The two most important are: whether the geometry of the conformers is optimized using an OPLS force field and the number of conformers the program is expected to generate. The default dielectric constant of 10 used during minimization was found to generate bioactive conformers better than using 1. A real-world example is shown in Figure 4. Apart from resolving bad atomic contacts, geometry optimization typically leads to mildly improved bioactive conformation recovery (see Table 1). Optimization, however, is about 20 times slower than running without minimization. Optimization also tends to reduce the number of conformers; something that can lead to reduced processing time in subsequent calculations that use these conformations.
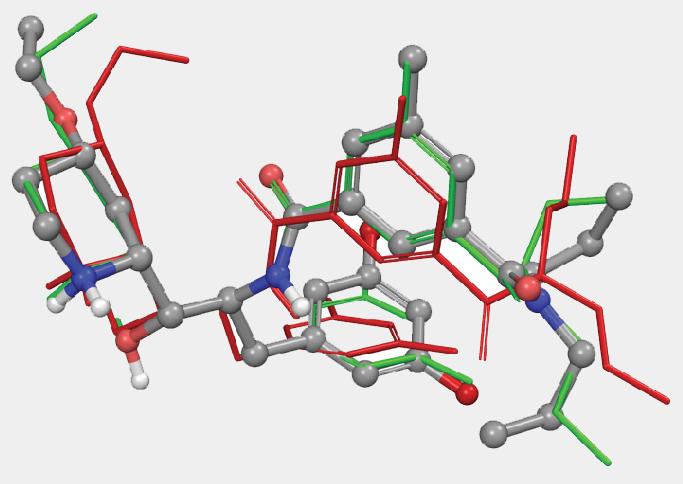
Another parameter controlling the speed/quality tradeoff is the number of conformers to be generated per input ligand (-n command line option): larger values of this parameter may often result in better conformer diversity. This is illustrated in Figure 4 for the ligand from 2QP8 PDB entry (1.5Å resolution): using default simulation settings, the conformer closest to the bioactive conformation has an RMSD of 1.3Å (red). The RMSD drops to 0.6Å (green) if the maximum number of conformers is increased from 64 (default) to 256, and geometry optimization using OPLS3 force-field is enabled.
Given its exceptional speed and ability to reproduce low RMSD bioactive conformations, ConfGen is the ideal solution for generating conformations for ligand-based virtual screening. It is tightly integrated within Maestro and Schrödinger’s ligand-based drug discovery tools, including Phase, which provides the ability to easily create databases for fast ligand-based virtual screening. As with all Schrödinger products, ConfGen is also constantly being refined and improved, so while the updates described in this paper represent a significant step forward compared to previous iterations, they will also act as the baseline for future enhancements.
J. Chem. Inf. Model,” 2017, 57(11), 2719-28.
J. Chem. Inf. Model., 2012, 52(7), 1757-68.
Chemistry, 2009, 15(1), 186-97.
Chemistry, 2009, 15(46), 12770-9.
Dalton Transactions, 2013, 42(24), 8617-36.
J. Phys. Chem., 1990, 94, 8897-8909.
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Access expert support, educational materials, and training resources designed for both novice and experienced users.
Shape-based methods for aligning and scoring ligands have proven to be valuable in the field of computer-aided drug design.1,2 Our Shape Screening tool is a powerful shape-based flexible ligand superposition and virtual screening method, which rapidly produces accurate 3D ligand alignments and efficiently enriches actives in virtual screening. Below, we describe the methodology, which is based on the principle of atom distribution triplets to rapidly define trial alignments, followed by refinement of top alignments to maximize the volume overlap. The method can be run in a shape-only mode or it can include atom types or pharmacophore feature encoding, the latter consistently producing the best results for database screening. We show that Shape Screening performs well in database screening calculations when compared with other shape-based methods, including ROCS, using a common set of actives and decoys from the literature. Three key considerations were emphasized when developing Shape Screening:
Shape Similarity Models
The basic concept of shape similarity is illustrated in Figure 1. Given a superposition of two structures A and B, the shared or jointly occupied volume VA∩B is normalized by the total volume VA∪B to arrive at a shape similarity SimAB that ranges between 0 and 1:
SimAB = VA∩B / VA∪B
A basic goal of shape screening is to determine the alignment of A and B that maximizes SimAB. The complexity of this task depends upon the mathematical representation of shape, and the way in which volumes are calculated.
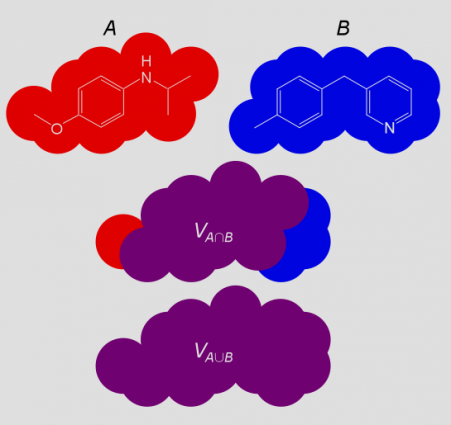
Shape Screening represents a structure as a set of hard atomic van der Waals spheres, with one sphere for each heavy atom and polar hydrogen. The overlap OAB between structures A and B is computed as the sum of pairwise atomic overlaps, and it is normalized by the largest self-overlap to obtain the following measure of shape similarity:
SimAB = OAB/max(OAA, OBB)
This differs from the previous definition in that an alternate normalization scheme is employed, and rigorously computed volumes are replaced by overlaps that ignore the effects of intersections among three or more atoms. Although ignoring higher order overlaps results in an overestimation of the true volumes, normalization by the largest self-overlap computed in the same manner tends to cancel errors, as shown in Figure 2. These approximations allow exceedingly fast shape similarity calculations compared to Gaussian-based methods,3,4 and the use of hard spheres eliminates the need to consider overlap between pairs of atoms separated by more than the sum of their van der Waals radii.
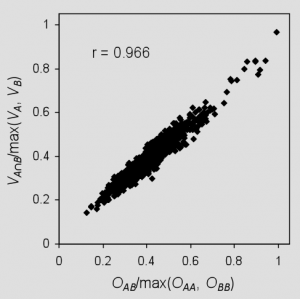
When computing overlaps, Shape Screening has the ability to treat all atoms equivalently, a so-called “pure shape” approach, or to distinguish atoms by type and consider overlap only between atoms of the same type. In the latter case, Shape Screening provides progressively more specific schemes that differentiate by Phase QSAR atom type,5 by element, and by MacroModel atom type.
As an alternative to the atom-based approach, Shape Screening can represent a structure as a set of pharmacophore sites that encode the locations of hydrogen bond acceptors and donors, hydrophobic regions, positive and negative ionizable functions, and aromatic rings. No particular pharmacophore model is implied by this approach, since all sites in a given structure are encoded into the shape, not just those that are hypothesized to be required for binding to a particular target. Pharmacophore sites are mapped to a structure using Phase feature definitions,5 and each site is represented by a 2 Ǻ hard sphere. Figure 3 illustrates the various models of shape that are supported.
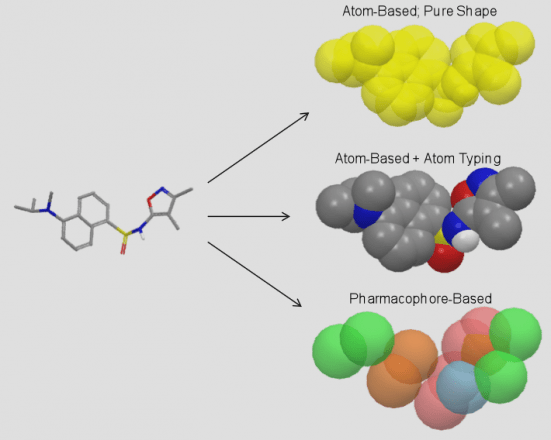
Whether an atom-based or pharmacophore-based approach is used, Shape Screening identifies numerous pairs of triplets with similar geometries and similar local environments in structures A and B and superimposes the two structures based on a least-squares alignment of each pair of triplets (Figure 4). The superposition with the highest shape similarity is then refined by realigning on additional pairs of atoms/sites that lie within 0.5 Ǻ of each other in the triplet-based alignment.
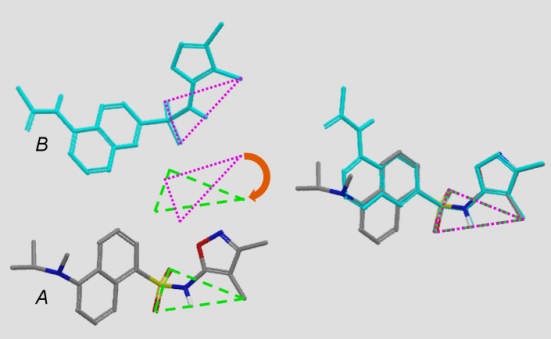
For each pair of structures, hundreds of alignments may ultimately be considered in a tiny fraction of a second. This is possible thanks to an optimized triplet alignment algorithm, ultra-fast hard sphere overlap calculations, and a shape similarity estimation technique which allows poorer overlays to be rejected after computing only a fraction of the total overlap. These time-saving measures allow Shape Screening to screen a multi-conformer Phase database at a rate of about 600 conformers per second on a 2 GHz processor. Shape Screening calculations are trivially parallelizable, and any desired speedup is achievable by dividing the screen over multiple processors.
Shape Screening Applications
Figure 5 illustrates the quality of overlays that can be achieved with Shape Screening using elemental atom types. Here, the CDK2 X-ray ligand structure 2G9X was used as a rigid template onto which nine other CDK2 ligands were aligned. Results are reported for the highest scoring X‑ray to template alignment, and for the highest scoring conformer to template alignment, where conformational ensembles were generated using both MacroModel and Shape Screening on‑the‑fly ConfGen sampling. In all cases, Shape Screening yields a multi-ligand alignment with low average RMSD values, and clean superposition of common structural elements.
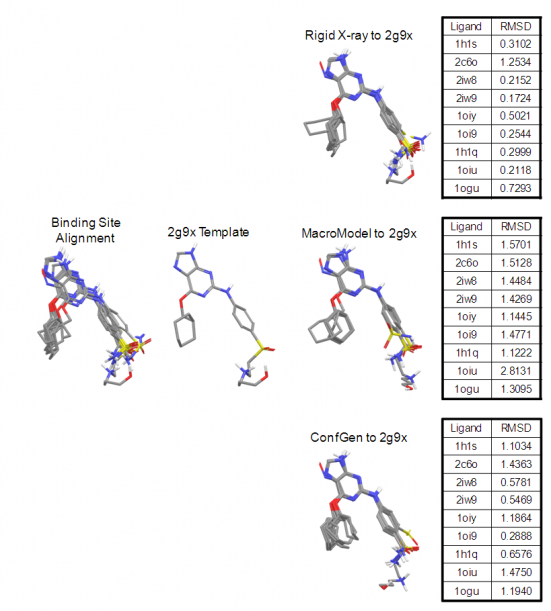
In addition to producing intuitive, high-quality overlays, Shape Screening has been shown to be quite effective at selectively identifying known actives within a database of drug‑like decoys.6 Table 1 summarizes results of Shape Screening virtual screening exercises performed according to the protocols described by McGaughey et al.7 Briefly, multi‑conformer actives for 11 diverse targets were seeded within a multi‑conformer database of 25,000 MDDR decoys. A single active for each target was used as a rigid template for shape-based screening, and database structures were ranked in order of decreasing similarity to that template.
As evidenced by the average enrichment factors in the top 1% of the screened database, results consistently improve with the use of more specific atom typing schemes. Analogous behavior was observed when 2D fingerprint screens were performed on the same data,8 so the relationship between atom type specificity and enrichment is not surprising. Although this trend is promising, improvements for most targets are only incremental, and it is unlikely that devising ever-more discriminating atom typing schemes will lead to a true breakthrough in performance. This threshold for performance improvement is not crossed until the atom-based model of shape is replaced with a pharmacophoric representation. Doing so boosts enrichments for eight of 11 targets, including a two‑fold or greater increase in four cases, and a 66% improvement over MacroModel atom types on average.
| Target | EF(1%) | ||||
|---|---|---|---|---|---|
| Pure Shape | QSAR | Element | MMod | Schrödinger Shape Screening | |
| CA | 10.0 | 25.0 | 27.5 | 32.5 | 32.5 |
| CDK2 | 16.9 | 20.8 | 20.8 | 23.4 | 19.5 |
| COX2 | 21.4 | 19.1 | 16.7 | 19.5 | 21.0 |
| DHFR | 7.7 | 3.9 | 11.5 | 23.1 | 80.8 |
| ER | 9.5 | 17.6 | 17.6 | 13.5 | 28.4 |
| HIV-PR | 13.2 | 17.7 | 19.1 | 14.0 | 16.9 |
| HIV-RT | 2.7 | 2.0 | 4.7 | 4.7 | 2.0 |
| Neuraminidase | 16.7 | 16.7 | 16.7 | 16.7 | 25.0 |
| PTP1B | 12.5 | 12.5 | 12.5 | 12.5 | 50.0 |
| Thrombin | 1.5 | 4.0 | 4.5 | 8.5 | 28.0 |
| TS | 19.4 | 32.3 | 35.5 | 51.7 | 61.3 |
| Average | 11.9 | 15.6 | 17.0 | 20.0 | 33.2 |
| Median | 12.5 | 17.6 | 16.7 | 16.7 | 28.0 |
Table 1: The enrichment factors at 1% screened for various Shape Screening approaches performed according to protocols described by McGaughey et al.7 Increasingly specific atom typing schemes are shown from left to right.
The pharmacophore-based approach also competes very well with other 3D virtual screening methods which have been applied to the McGaughey data set. Table 2 compares Shape Screening pharmacophore-based enrichments to those obtained using the ROCS-color technique9 and the SQW superposition method developed at Merck.7,10 Shape Screening surpasses both of these methods by 30-40% in terms of average and median enrichments, and outperforms each of them head-to-head in eight of 11 cases. Since publication of the McGaughey paper in 2007, ROCS‑color has been viewed by many as the gold standard for shape-based screening, so these latest results are of particular significance.
| Target | EF(1%) | ||
|---|---|---|---|
| Schrödinger Shape Screening | SQW | ROCS-Color | |
| CA | 32.5 | 6.3 | 31.4 |
| CDK2 | 19.5 | 9.1 | 18.2 |
| COX2 | 21.0 | 11.3 | 25.4 |
| DHFR | 80.8 | 46.3 | 38.6 |
| ER | 28,4 | 23.0 | 21.7 |
| HIV-PR | 16.9 | 5.9 | 12.5 |
| HIV-RT | 2.0 | 5.4 | 2.0 |
| Neuraminidase | 25.0 | 25.1 | 92.0 |
| PTP1B | 50.0 | 50.2 | 12.5 |
| Thrombin | 28.0 | 27.1 | 21.1 |
| TS | 61.3 | 48.5 | 6.5 |
| Average | 33.2 | 23.5 | 25.6 |
| Median | 28.0 | 23.0 | 21.1 |
Table 2: A comparison of the Shape Screening pharmacophore-based approach to other 3D virtual screening methods.
Other versatile features of Shape Screening include the ability to score poses in place, force the alignment of specific atoms by way of SMARTS matching, compute similarities to multiple shape queries in a single run, apply alternate similarity normalization schemes that facilitate the identification of embedded shapes, and filter hits using excluded volumes. The Shape Screening technology has also been employed to develop a fast, multi-ligand superposition method, where the template and the structures being aligned to it are all treated in a flexible manner, and the template conformer that yields the best overall alignment of all ligands is utilized.
References
In collaboration with Janssen and Nimbus, Schrödinger published a preprint in ChemRxiv, A Free Energy Perturbation Approach to Estimate the Intrinsic Solubilities of Drug-like Small Molecules. The paper presents a novel physics-based approach to predicting the aqueous solubility of small molecules that takes into account three-dimensional solid-state characteristics in addition to polarity. The method performed well not only on public datasets, but in testing against the measured solubilities from a large number of advanced programs in-house at Janssen. Furthermore, the paper reports on the first prospective application of the method in a live drug discovery project in collaboration with Nimbus Therapeutics.
Bringing together FEP+ Solubility, FEP+ potency, RRCK passive permeability modeling, and physico-chemical parameters like PSA in a modeling screening funnel can substantially improve the probability of success and accelerate the project timeline.

Dr. Mondal explains the paper’s key findings:
Tell us what’s the key advance in this paper, and why is it important?
In this paper, we introduce a brand new technology, FEP+ Solubility—a physics-based method that provides a detailed examination of the 3D solid-state packing structure of a molecule and accurately predicts solubility without a training set. FEP+ Solubility is built on our free energy perturbation method, FEP+, and is both accurate and has a wide domain of applicability, making it particularly suitable for drug discovery research, where exploring novel chemical space could be crucial to success.
Aqueous solubility is a focal point during lead optimization in drug discovery because it affects bioavailability, a molecule’s ability to enter the bloodstream and circulate throughout the body. Given that it is estimated that almost 40% of all new chemical entities (NCE) are determined to be insoluble, or nearly so, and that molecules with low solubility often experience issues during clinical and pre-clinical trials, leading to failure, it becomes clear why improving solubility is often a focal point during lead optimization. Moreover, straightforward attempts to increase solubility during lead optimization can degrade membrane permeability and/or potency.
How did researchers traditionally try to improve solubility? And how does FEP+ Solubility compare with previous approaches?
The most common approach to improving solubility was empirical—guided usually by logP or sometimes by machine learning models fitting existing data of experimentally measured solubility to various molecular descriptors, such as logP and polar surface area. However, such empirical approach can actually lead one astray, for example, simply adding polar groups can preferentially stabilize the solid state, and lead to the sometimes counterintuitive result of reducing solubility. FEP+ Solubility, on the other hand, allows for a detailed computation of the 3D solid-state packing energetics, providing insightful predictions about interactions that can improve or degrade solubility. A great example of this is Benzodiazepine, where it has been shown that substitutions that disturb favorable solid-state interactions can lead to improved solubility.
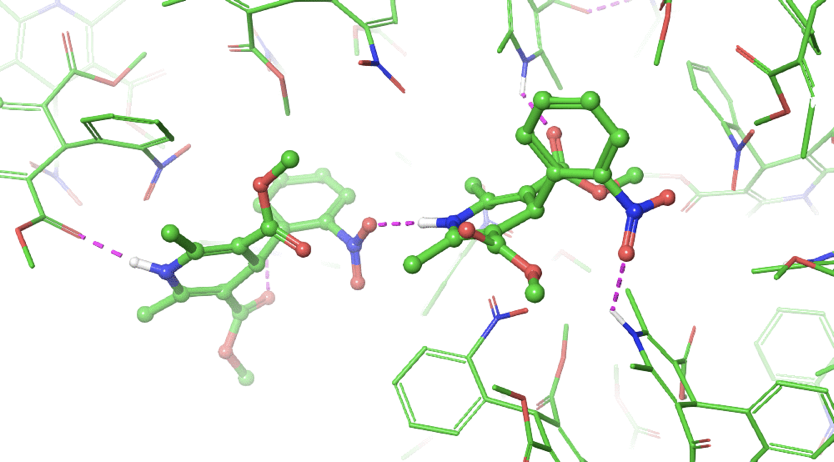
Another key difference between empirical methods and FEP+ Solubility is that the former requires a training set, and thus is inherently limited in its applicability to the chemical space of the training set, and often struggles to extrapolate to novel chemical entities. Unfortunately, it’s often necessary to expand beyond this limited scope of chemical space in order to achieve the desired solubility in balance with other parameters or IP, and this is where FEP+ Solubility really shines with its large domain of applicability. Furthermore, the understanding gained from FEP+ Solubility studies such as elucidating an in silico solubility SAR, can help the project team ideate novel chemical structures to improve solubility.
Can you speak to the performance of this new method, FEP+ Solubility, and how you are using the technology?
Sure, we did extensive retrospective studies to test our model against available published solubility data for compounds of pharmaceutical interest, and saw great agreement. But more importantly, we began to use the model in prospective studies, and again we saw excellent performance by FEP+ Solubility in classifying compounds based on their solubility profile, making it a quotidian tool in our arsenal.
As part of Schrödinger’s Drug Discovery Group, we use the Schrödinger platform in all of our internal and collaborative drug discovery projects. The platform consists of three things: advanced physics-based methods, machine learning, and enterprise informatics.
The predictions from various physics-based methods including FEP+ Solubility and machine learning methods, where applicable, are captured within LiveDesign, our web-based enterprise informatics program, facilitating synthesis decisions based on a holistic comparison of a large number of ideated molecules. The most promising compounds emerging from each round are then further optimized through additional cycles of computational analysis enabling us to determine the best candidates for synthesis.
FEP+ Solubility sounds like a significant advancement, how do you see it impact drug discovery projects?
Drug discovery teams often find themselves trapped in lengthy and uncertain cycles of optimization where they change R-groups to improve solubility, only to lose ground against potency, or an ADME property like permeability; or vice versa. These parameters need to be satisfied in the same molecule but are generally anti-correlated, meaning that if we are limited to designing along the dimension of polarity / hydrophobicity alone, often it would be to the detriment of other important properties.
By going beyond just polarity, it becomes easier to overcome the potency / selectivity / permeability / solubility tradeoffs to obtain higher quality chemical matter faster. With FEP+ Solubility used in conjunction with FEP+ analyses of potency and selectivity, and RRCK passive permeability modeling at scale, it becomes possible to break out of insoluble regimes and achieve balanced profiles within 1-2 cycles of synthesis.
This is especially true for projects where most synthesized compounds are found to be insoluble, the probability of success for meeting the Development Candidate guidelines can be dramatically improved by focusing synthesis on high quality chemical matter predicted to be soluble, potent, selective, and permeable.
The value of using FEP+ for solubility is particularly high when using it together with FEP+ for potency and selectivity, and RRCK permeability modeling, because the net outcome is more than the sum of the individual analysis. For example, one may decide to remove an aromatic ring based on FEP solubility results; however, if the aromatic ring is productive in the binding site, the probability of success is much higher if FEP+ potency modeling is used to maintain the potency while replacing the aromatic ring.
Our modeling platform allows researchers to map out the SAR for potency, selectivity, permeability, and solubility side-by-side for different series and sub-series. This information can help teams strategize which core to prioritize for further optimization, how to functionalize a given core, and to stimulate the iterative design of new ideas.
Any final thoughts?
To summarize, our approach through the FEP+ framework provides an opportunity to systematically predict the solubility of novel molecular entities. Beyond early stage drug discovery, this work lays down the foundation for solving key problems in formulation and materials science contexts with computational modeling. That is a world of impactful advances waiting to be made!

Dr. Andrea Browning
Scientific Lead, Polymers and Soft Matter
Materials-focused research and development must address not only challenges for improved functionality but also emergent issues for existing products. These engineering-level problems frequently have a molecular root cause. Experimental and simulation techniques probing the molecular level provide critical information for solving many of these problems, but relating them to the engineering level can be challenging. This webcast will review case studies where this connection has been made and leveraged with molecular simulation in consumer packaging and adhesive materials.
We sat down with Dr. Jianxin Duan to discuss his paper, “Improving the Accuracy of Protein Thermostability Predictions for Single Point Mutations” which recently appeared in the Biophysical Journal.
Tell us what was the main motivation behind this study?
The function of a protein is tightly coupled with its structure and dynamic behavior. Therefore, understanding the thermostability of proteins can provide fundamental insights into how they work. For example, single point missense mutations may directly affect protein function, leading to diseases. Many of these mutations have been found to be linked to protein thermostability. Also, from a practical perspective, protein stability engineering has wide applications in various industries, for example, vaccines and antibodies need to be engineered to have long shelf life and prevent aggregation; industrial enzymes for food, detergents, paper, or fuel need to be designed to be stable and functional in the desired environments.
As you know, we’ve had tremendous success in using our FEP+ technology in advancing small molecule drug discovery—we’ve published many papers, and all of our drug discovery projects have achieved milestones at an accelerated pace when compared to traditional approaches. But what’s less well established is how well FEP+ could advance projects involving biologics, so we set out to design a study to specifically examine the performance of FEP+ in predicting protein thermostability. We aimed to assess both quantitatively and qualitatively the technology as a viable option to predict changes in protein thermostability upon mutations such that it may guide protein engineering projects.
Had there been previous studies using FEP on proteins? And if so, how does the current study differ?
The idea of applying free energy perturbation theory (FEP) to predict the effect of single point mutations on protein stability is not new; however, production-level calculations to predict protein stability in industrial settings have not been possible due to a couple of severe limitations, including the inability to model mutations to and from prolines, where the bonded topology of the backbone is modified, and the complexity in modeling charge-changing mutations. In this study, we extended the FEP+ protocol to accurately model both of these types of mutations. Furthermore, because FEP calculations could be computationally intensive, many earlier studies were limited to very short simulations, and with limited solvation around the mutation, ultimately resulting in less accurate predictions. Thanks to algorithmic improvements and hardware advances in GPUs, Schrödinger’s FEP+ has demonstrated repeatedly that it can meaningfully impact projects within realistic timelines.
Because we set out to specifically assess FEP+’s performance in dealing with the challenging proline and charge-changing mutations, we paid particular care to selecting the test systems so that we could quantitatively evaluate our results – we curated a data set with high-resolution crystal structures for both wild-type and mutants, and with accurately measured stability data. You may find details of our test systems in the paper. And I’m happy to report that we achieved comparable accuracy in predicted stability changes as observed previously in countless small molecules studies, demonstrating that FEP+ is indeed an appropriate computational strategy for studying biologics.
Are there other methods for predicting protein thermostability? And if so, how do the present FEP+ results compare?
Yes, because of the importance of being able to accurately predict protein stability, many different methods have been proposed over the years. We compared our FEP+ results with several of the more popular methods.
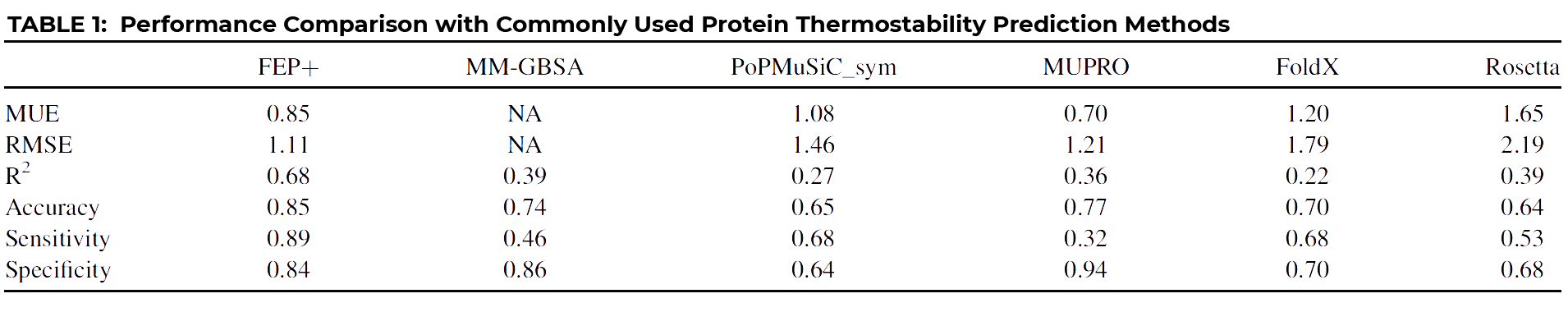
As you can see at a glance, FEP+ outperforms all other common approaches quantitatively, with the possible exception of MUPRO, which is a machine learning model using only sequence information and all mutations as part of its training set. However, it’s been suggested that MUPRO may suffer from training set bias, and indeed this is born out when you examine the qualitative measures of Accuracy, Sensitivity, and Specificity, also given in the table.
Let me explain what these three measures mean by using an example:
In order to have a truly useful classification tool, you’d want a method that scores consistently high across all three indicators, like FEP+, which clearly outperforms all other methods, including MUPRO, which while scoring well for Specificity, owing to its training set, it nevertheless is biased by that training set to be a poor predictor of stabilizing mutations, as indicated by its very low Sensitivity score of 0.32.
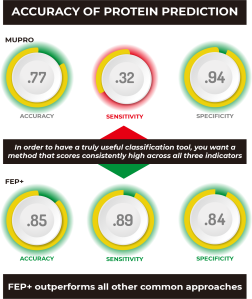
But isn’t FEP+ computationally expensive? How well can it be deployed at scale to meaningfully accelerate a discovery program?
It is true that FEP+ studies require a computational investment, but thanks to software algorithmic improvements and the ever-increasing processing power of GPU hardware, full analyses using FEP+ is well within reach for real-life projects. Furthermore, as described in our paper, we devised a screening cascade taking advantage of the much more computationally efficient residue scanning, available within our BioLuminate application, which tends to err on the side of false positives, as a filter for further FEP+ analyses. This way, one could rapidly screen all relevant mutations, and only pass onto FEP+ those that are found to be stabilizing, and let FEP+ identify those mutations that are most likely to be true positives.
That all sounds very promising, are there future plans for enhancing or expanding the technology?
Yes, of course, we’ve learned a great deal from this study, such as how to best model the protein’s unfolded state to achieve higher accuracy in the predicted change in stability. We also scrutinized our results and identified potential cases that may require longer simulations to fully capture large structural changes. We will continue to refine the protocol to improve the performance both in terms of prediction throughput and accuracy.

Director, Applications Science, Schrödinger
Jianxin Duan, Ph.D., is a Director with Schrödinger’s Applications Science group. He earned his Ph.D. in Biophysics from Karolinska Institute in Stockholm. Dr. Duan has been with Schrödinger for 14 years. He was involved in the development of ligand based methods, including cheminformatics, pharmacophore modelling and machine learning. His current personal research focuses on protein and antibody design.