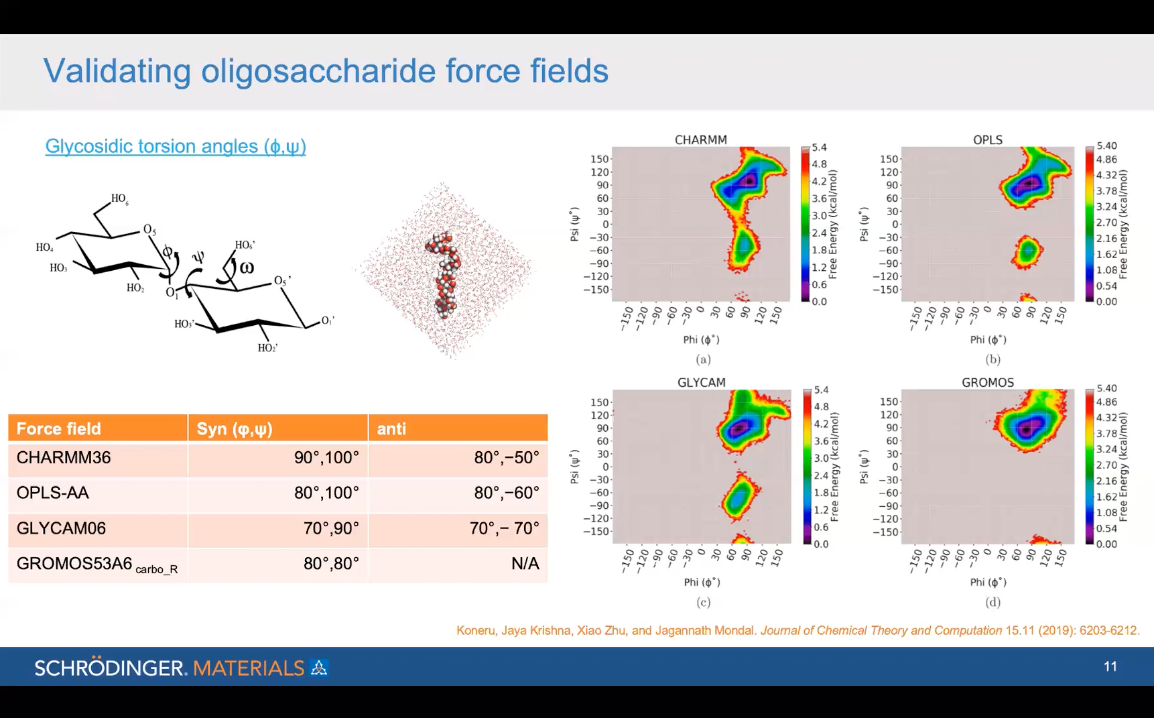
Advancing the understanding of the starch structure-function relationship with molecular simulation
Share
Speaker:
Jeffrey Sanders, Principal Scientist
Abstract:
Starch is one of the most common biopolymers in food products and it’s interaction with other ingredients influences the overall physical and chemical characteristics, including nutrition and texture quality. Given the relative size of and complexity of starch polymers, traditional biophysical experiments lack the necessary resolution to study key nanostructure properties. Use of molecular dynamics simulations provides critical insight into the structural and dynamic properties of amorphous starch structures. Combining structural analyses with grand canonical Monte Carlo methods, the moisture uptake behavior is also explored along with it’s effects on thermophysical properties.
With the advent of more powerful hardware and methods, the use of machine learning (ML) methods has seen a significant upsurge in chemistry-related applications recently. Specifically in drug discovery, the prediction of ADMET (absorption, distribution, metabolism, excretion and toxicity) properties is a main target for ML applications. Herein, we present performance metrics for Schrödingers automated ML model building engine, DeepAutoQSAR, on the ADMET subset of the Therapeutic Data Commons (TDC) — a large collection of public data for ML model building and benchmarking. We also compare the performance of DeepAutoQSAR to the performance of two open source projects, namely ChemProp and DeepPurpose.
DeepAutoQSAR is among the top-performing methods in 20 of the 22 investigated cases, clearly outperforming the other methods in 9 of those. For the other 11 cases, at least one of the other tested methods performs similarly. We believe that continuous development and further improvement of DeepAutoQSAR, in accuracy, robustness to chemical data shift and label efficiency will enable faster and more cost-effective means of drug discovery, ultimately leading to the introduction of novel therapeutics.
Introduction
It is widely recognized that the ADMET (absorption, distribution, metabolism, excretion and toxicity) profile of novel molecules plays a key role in the successful development of new drugs. This is reinforced by the amount of time and effort spent both in academia and the pharmaceutical industry to develop reliable models to measure and predict numerous related endpoints1. Due to the potentially catastrophic impact of an unfavorable ADMET profile in the later stages of drug development, a common goal is to identify potential issues as early as possible.
With the rise of ultra-large on-demand libraries and DNA encoded libraries (for example Enamine REAL Space or WuXi LabNetwork), early identification of liabilities requires methods that are computationally fast, cheap, and accurate enough to evaluate hundreds of millions of compounds without discarding potentially good candidates. This obviously precludes the use of experimental in vivo or even in vitro methods. Modern machine learning (ML) approaches, often coined artificial intelligence (AI), can easily process millions of molecules on short timescales and low computational costs with acceptable accuracy.
In contrast to physics-based in silico methods, ML/AI methods require high fidelity data to be trained to predict a given endpoint. High-quality training data is often unavailable; data need to be clean and well-curated, and datasets in chemistry applications are often smaller than those used in other domains like ML on images or text. These strict data requirements can limit the application of more complex ML/AI approaches since there is often insufficient amounts of training data to fit complex and accurate models.
However, recognizing the importance of profiling ADMET properties over the past decades, large pharmaceutical companies have generated a wealth of data which is often unfortunately non-public and exclusively applied for internal programs. Public data is rarer, but there are efforts to collect and aggregate public data 2 and also to share non-public data in smart ways to improve existing models while retaining data confidentiality 3.
The successes of deep learning (DL) approaches have led to a renaissance of ML/AI in chemistry applications, with a large number of both open-source and commercial software to pick from when targeting ADMET endpoints. While open-source software oftentimes can profit from faster development cycles and thus implements new scientific insights more quickly, application is often limited to domain experts. On the other hand, commercial software has the benefits of structured quality assurance (QA), documentation and support, and comes coupled with comprehensive user interfaces which significantly lower the barrier to entry for non-experts.
In this paper, we will take a closer look at the performance of two of the more popular open-source packages, ChemProp and DeepPurpose, and Schrödinger’s ML/AI package DeepAutoQSAR, demonstrating their comparative performance on a recently published set of benchmarks.
Hit to lead design of novel d-amino-acid oxidase inhibitors using a comprehensive digital chemistry strategy
Computational platform grounded in highly accurate predictive models enables team-based discovery of a novel chemical series engaging a complex CNS target.
Inhibition of D-amino-acid oxidase (DAO) has been hypothesized as a potential therapeutic strategy for schizophrenia. Schrödinger’s Drug Discovery Team engaged in a discovery effort with a collaborator to identify novel DAO inhibitors with potential best-in-class properties.
Program Challenges
Identify novel chemical matter while striving for best-in-class molecules that cross the blood-brain-barrier
Simultaneously optimize drug-like properties, improve CNS exposure, and affinity
Approach
The Drug Discovery Team deployed a large-scale digital chemistry strategy leveraging:
A centralized project data platform to facilitate knowledge-based medicinal chemistry design collaboration (LiveDesign, AutoQSAR)
Physics-based methods to predict affinity and prioritize design ideas for synthesis (FEP+)
Computationally-driven ideation and scoring workflow to amplify common enumeration strategies and screen hundreds of millions of compounds using machine learning coupled with physics-based free energy methods (FEP+, AutoDesigner)
Results
The team discovered a novel class of DAO inhibitors with desirable drug-like properties by confidently exploring synthetically-challenging chemistry. The team also identified a previously unexplored subpocket for further evaluation. The novelty of the compounds, coupled with well-balanced properties, demonstrates the extraordinary power of the approach to unleash project team creativity. By leveraging a digital platform, the team explored vast chemical space while simultaneously optimizing for drug-like properties in a challenging disease area.
Why use a digital chemistry approach?
A digital chemistry approach uses physics-based modelling, machine learning, and a team-based collective intelligence platform to design better molecules on accelerated timelines.
How to achieve optimal drug-like properties?
The development of CNS drugs poses several unique challenges. Fine-tuning physicochemical properties for optimal brain exposure is an essential element of CNS drug development. Many companies have discontinued neuroscience discovery because these challenges lead to longer development timelines and a lower probability of success.
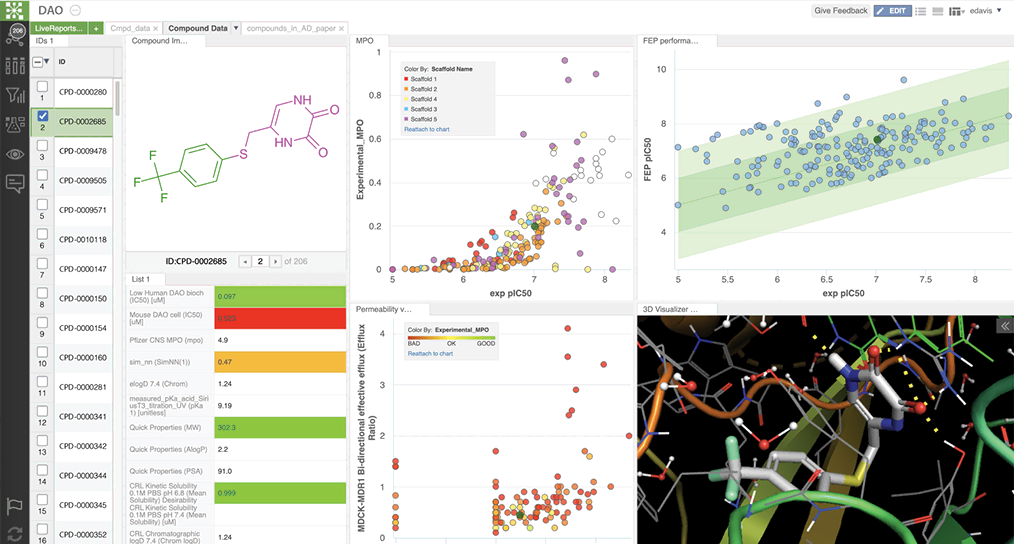
Schrödinger’s Drug Discovery Group developed property prediction models, using AutoQSAR deployed via LiveDesign, a web-based collaborative design platform. LiveDesign enabled teams of medicinal chemists to crowdsource designs and simultaneously optimize CNS properties with push-button workflows in a single interface (see figure 1). Compounds predicted in the desired property space were triaged using free energy perturbation (FEP+), a physics-based method for accurately predicting compound binding affinity. This workflow empowers teams to confidently pursue synthetically challenging compounds.
Figure 1. Schrödinger’s digital collaboration platform, LiveDesign, facilitates design optimization through custom multiple parameter optimization models by centralizing program data and improving team communication and collaboration.
How did a digital chemistry strategy enable improved hypothesis testing?
The team delivered high-quality molecules by working in an ecosystem that facilitated exploration of vast, novel chemical space and simultaneous optimization for desired properties through accurate physics-based modeling and machine learning.
The digital chemistry approach allowed the team to discover and quickly overcome many medicinal chemistry challenges in the pursuit of best-in-class molecules (see figure 1).2
Figure 2. SAR progression to achieve key milestones through late lead optimization with key compounds series represented. Crucial discovery and medicinal chemistry outcomes are highlighted. DHP represents dihydropyrazine and NHP, N-hydroxyl pyrimidine.
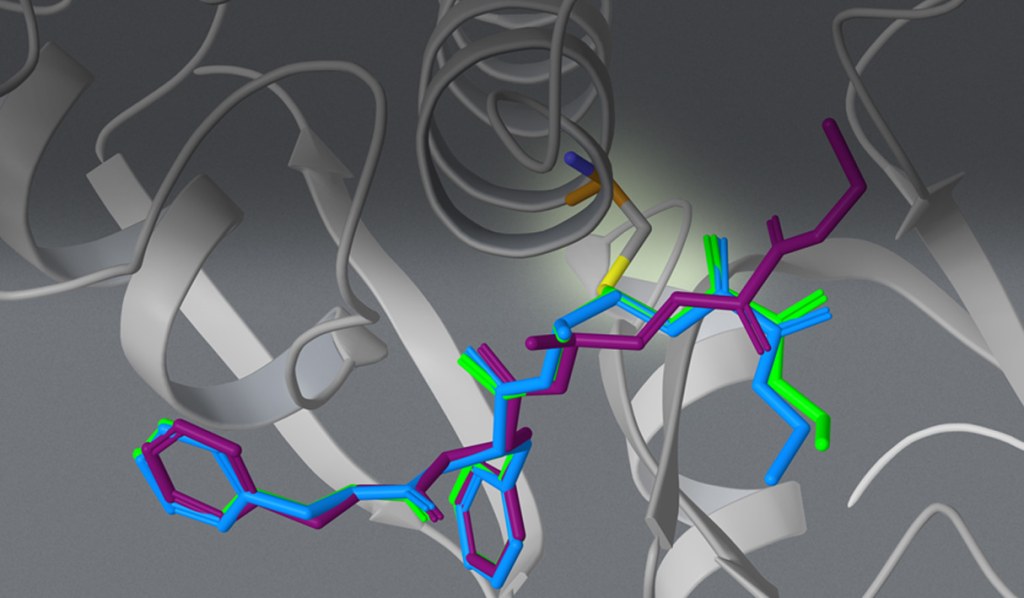
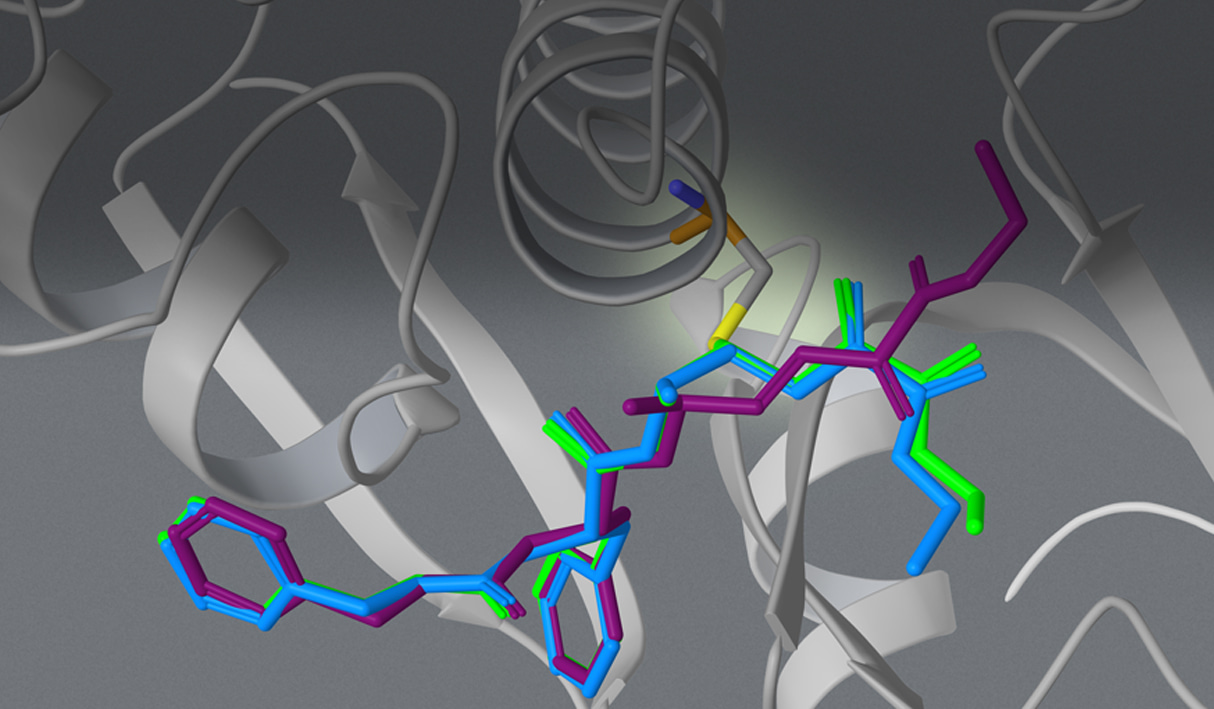
The team interrogated the atypical polarity of the DAO binding site, shown in panel A of figure 3. Chemists pursued challenging chemistry to reduce conformational flexibility and displace a high-energy water molecule by cyclizing and methylating cmpd 4 and 5 (see figure 2, panels B and C).
Finally, while literature and crystallographic structures suggested limited pocket volume for SAR exploration,3 FEP+ revealed the opportunity to interrogate this vector with larger chemical groups such as cmpd 6, as shown in panel D of figure 3.
Figure 3. A) Polarity of DAO binding site required a polar warhead. B) Cyclization of ligand linker reduced entropy improving affinity C) Displacement of high energy water near the binding site improved affinity (compare water present in panel B with panel C). D) FEP+ suggested exploring a novel subpocket predicted to improve potency (compare gray surface in panel A with the green surface in panel D).
How to interrogate vast chemical space and deliver novel chemical matter?
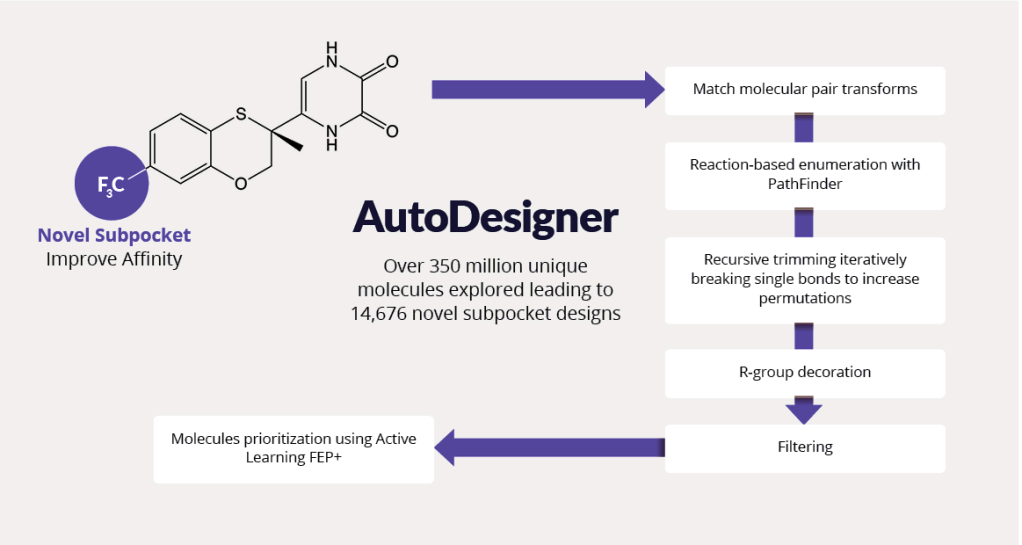
Pursuing best-in-class molecules required exploring vast chemical space outside of previously characterized drug-like molecules. The team utilized AutoDesigner, a multifaceted large-scale enumeration workflow (figure 4), to generate ideas exploring the novel DAO binding subpocket suggested by FEP+ (see figure 3, panel D).4
Figure 4. AutoDesigner enumeration and triage workflow explores SAR from the lead molecule while optimizing CNS drug-like properties to discover best-in-class molecules by covering vast chemical space.
To explore SAR and tune physicochemical properties, the team performed iterative cycles of AutoDesigner in the newly discovered subpocket. After triaging with appropriate filters, all molecular ideas were prioritized using free energy methods. The team trained active learning models using physics-based affinity predictions (FEP+) to prioritize compounds for synthesis. In total, more than 350 million ideas were generated and triaged.
What was the project impact?
Typically as drug discovery programs progress, teams struggle to balance desired properties, which leads to deficits in desired drug properties as novel scaffolds are explored and optimized.
Through a computational platform rooted in creative team collaboration, highly accurate predictive modeling, and enhanced by machine learning, a promising CNS DAO inhibitor series transitioned from hit discovery to lead optimization with approximately 11,000 compounds scored by FEP+ and only 208 synthesized. Of the 208 compounds synthesized, only 20 were inactive (>10μM) against DAO. By discovering novel compounds and concurrently performing multi-parameter optimization of critical CNS properties, the team delivered high project impact for this challenging disease area.
References
Bromet E.J., Fenning S.; Epidemiology and natural history of schizophrenia. Biol Psychiatry. 1999, 46 (7), 871–881.
Tang et al. Discovery of a Novel Class of D-amino Acid Oxidase (DAO) Inhibitors with the Schrödinger Computational Platform. ChemRxiv. Preprint. https://doi.org/10.33774/chemrxiv-2021-dkf1k.
Hondo et. al. 4-Hydroxypyridazin-3(2H)-one Derivatives as Novel d-Amino Acid Oxidase Inhibitors. J Med Chem. 2013, 56 (9): 3582-3592.
Bos et al. AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of D-Amino Acid Oxidase Inhibitors. ChemRxiv. Preprint.
With the recent resurgence in covalent drug research, computational insight into covalent docking is becoming key to understanding how covalent inhibitors can be used to address selectivity and potency challenges.
Covalent inhibitors derive their activity not only from the formation of a covalent bond between the target and the ligand but also from stabilizing non-covalent forces in the binding pocket. CovDock selects the top covalent complexes using the extensively validated Prime energy model, and calculates an apparent affinity score that captures these essential elements of a successful covalent docking process:
The pre-reactive ligand form occupies the binding pocket with enough residency time to facilitate the reaction of the ligand warhead with the reactive protein residue; and
unfavorable steric clashes and poor electrostatic contacts are prevented as the reaction proceeds.
CovDock begins with Glide docking to a receptor with the reactive residue trimmed to alanine. The receptor reactive residue is then added and sampled to form a covalent bond with the ligand in different poses. Covalent complexes are minimized using the Prime VSGB2.0 energy model to score the top covalent complexes. An apparent affinity score, based on the Glide score of pre-reactive and post-reactive poses, is also calculated to estimate binding energies for use in virtual screening.
Features
Accurate binding mode prediction:
CovDock is built upon a foundation of the time-tested Glide docking algorithm and Prime structure refinement methodology for accurate prediction of non-covalently docked poses. Glide quickly samples a large pool of initial poses for the pre-reactive species and Prime simultaneously optimizes the ligand pose and attachment residue to produce a sound physical chemistry. The resultant accuracy outperforms other docking programs in achieving lower RMS deviations from native co-crystallized structures.
Complete workflow:
CovDock performs a series of automated steps based on a simple setup from the Maestro graphical interface or from the command line. First, CovDock docks the pre-reactive ligand to determine viable poses that bring the reactive group into close proximity with the reactive receptor residue. Then the covalent bond is formed for the top scoring complex structures, the covalently attached ligand is sampled, and the complexes are scored using all-atom molecular mechanics with the OPLS force field and VSGB2.0 implicit solvent model.
Intuitive graphical interface:
Schrödinger’s intuitive graphical user interface, Maestro, provides easy-to-use panels for straightforward set-up of experiments, easy visualization, and efficient analysis of CovDock results.
Covalent Reactions Repository
Schrödinger has made available several custom reactions that can be used in CovDock studies, which can be found on the Covalent Reactions Repository documentation page.
Publications
“Docking covalent inhibitors: A parameter free approach to pose prediction and scoring”
Expanding the Limits of in silico Macrocycle Design
Due to their great potential in many drug discovery projects, significant efforts have been made to explore both natural and synthetic drug-like molecules that contain macrocycles. In the past, macrocycle design has proved to be challenging both synthetically and computationally. Computationally, chemical software is not typically designed for large flexible rings, but Schrödinger software provides several macrocycle design solutions including the ability to perform sampling using Prime, predict permeability, and dock macrocycles using Glide. It also enables users to estimate ring strain as well as compare relative affinities between compounds using FEP+ making macrocycle design more accessible than ever before.
Features
Sampling with Prime:
Using technology adapted from hierarchical protein loop sampling, Prime macrocyle sampling for small macrocycles is:
Accurate – Median backbone RMSD of 0.40 Å, with few outliers over 1.5 Å
Diverse – Output conformations are unique
Fast – Median calculation times are 5-10 minutes
Scalable – Providing accurate and diverse results for cyclic peptides and macrocyclic natural products
Passive Membrane Permeability:
Permeability of macrocyclic and non-macrocyclic molecules can be predicted by approximating the transfer free energy of molecules between water and a membrane using macrocycle sampling and fast implicit solvent models.
Docking with Glide:
Integrating macrocycle ring sampling into Glide docking creates a protocol that is:
Accurate – 65% of cases within 2.0 Å RMSD for self-docking of macrocycles
Fast – About an hour, on average, per compound including ring sampling and docking
Ring Stability Prediction:
Our predictions of the macrocycle ring strain required to adopt conformations similar to that of a known active ligand can be used to screen linkers to find those that will result in high-affinity macrocyclic versions of linear ligands.
Free Energy Perturbation with FEP+:
FEP+ can compare the relative affinity of congeneric macrocyclic and non-macrocyclic compounds, even when the difference is in the ring or the creation of a ring.
Publications
“Automated Design of Macrocycles for Therapeutic Applications: From Small Molecules to Peptides and Proteins”
Optimizing Protein Stability Using New Computational Design Approaches for Biologics
Share
Speaker
Dr. Guido Scarabelli
Senior Scientist
Abstract
Physical stability is a key determinant of the clinical and commercial success of biological therapeutics, vaccines, diagnostics, enzymes, and other protein-based products. The development of accurate computational methods for predicting protein stability can reduce the cost and time of experimental mutant design campaigns. In this work, we demonstrate the success of a rigorous physics-based computational method, Free Energy Perturbation (FEP), at quantitatively evaluating the relative thermodynamic stability of a diverse set of proteins on a dataset consisting of 328 single point mutations spread across 14 distinct protein structures and we will discuss the effects of simulation conditions on the computational predictions.
A crucial process in manufacturing CPUs and other high-tech devices is the deposition of solid material from reactive vapors. Different precursor vapors are used for chemical vapor deposition, vapor phase epitaxy, atomic layer deposition – and indeed the reverse process of atomic layer etching – with the precursor chemistry carefully designed for each case so as to control material quality at the nanoscale. But what all these techniques have in common is that the precursor chemicals must evaporate or sublime at a low enough temperature. Too much heating when vaporizing a precursor can make it decompose, causing it to be undeliverable to the growing surface.
With volatility playing such a central role in this technology (and in other fields like distillation, refrigeration, inkjet printing, food and perfumes), it is surprising that we understand so little about it. Volatility is the product of a remarkably fine balance of interatomic forces, dictating the extent to which molecules condense together as a solid or liquid, or bounce apart into a vapor and deliver a certain vapor pressure at any given temperature. These interatomic forces can be computed very precisely with quantum mechanics for one molecule or a group of molecules, but not at the scale of a liquid or solid. Even with today’s computing power, routinely and accurately predicting precursor volatility ‘from first principles’ remains unfortunately out of reach.
Machine learning approaches
Could an alternative more empirical approach prove useful? Does enough experimental data exist to find the relation between volatility and chemical structure? The vaporization of some organic molecules, such as alcoholic fractions or natural fragrances, has been of interest for centuries and high-quality vapor pressure data are available in the literature. Over the last decade, these data have been analyzed with advanced fitting algorithms that come under the umbrella of ‘machine learning’. Schrödinger has leveraged the latest machine learning techniques to develop a highly-accurate model that predicts the volatility of organic molecules up to C20.
However, when building machine learning models to predict volatility of precursor molecules, which are typically organometallic complexes, the situation is not so straightforward. New precursor molecules are constantly being proposed and evaluated. Commercial sensitivity sometimes means that data are partially withheld, or plagued by experimental configuration differences from laboratory to laboratory. Additionally, for the common aim of material processing, complete pressure-temperature curves are rarely measured, as it is more pragmatic to focus on the temperature for vapor to transport successfully to the reactor. As a result, datasets for building predictive models are sparse and incomplete.
Prediction of volatility for inorganic and organometallic complexes
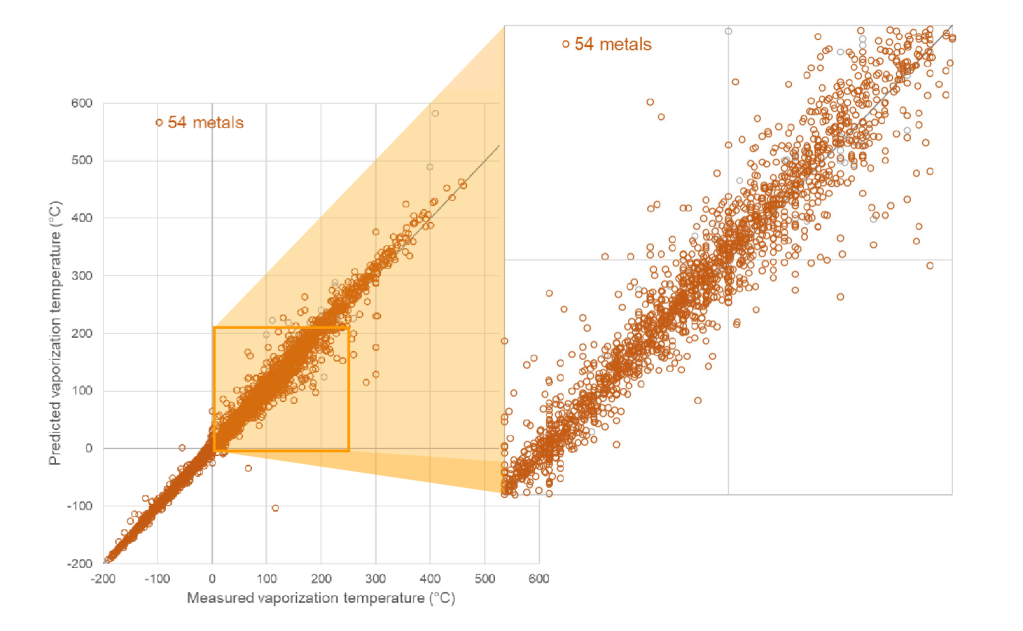
Schrödinger scientists embarked on the challenge of building machine learning models to predict the volatility of precursor molecules. Using in-house expertise in machine learning and advanced informatics, Schrödinger scientists collated and digitized information about organometallic precursors from disparate literature sources and applied a variety of machine learning algorithms (such as Random Forest and Neural Networks) in conjunction with different chemoinformatic descriptors and fingerprints. The result is the first capability of its kind for accurately and efficiently predicting the volatility for inorganic and organometallic complexes from their chemical structures. For complexes of the fifty most common metals and semimetals, the model predicts the evaporation or sublimation temperature at a given vapor pressure with an average accuracy of ±9°C (which is about 3% of the absolute temperature). As a trained model, the turnaround time is fast with the ability to compute hundreds of complexes per second.
New avenues for precursor development
This predictive model opens a new path for designing novel precursors with improved performance, not only improving their deposition or etch chemistry, but also optimizing the temperature at which they evaporate or sublime and can be delivered as a vapor. This advance will allow a much wider range of structural modifications to be screened computationally than before and will produce candidate precursors for experimental synthesis and testing that are both less risky and more innovative. This volatility model, together with Schrödinger’s quantum mechanics-based workflows for computation of reactivity and decomposition, gives scientists a complete design kit for vapor-phase deposition or etch, delivering a faster pace of research into materials and processes for new technologies.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Sublime Precursors: How Modelling Organometallics at Surfaces Drives Innovation in Materials Processing
Share
Speaker
Simon Elliott
Director- Atomic level process simulation
Abstract
The rich and often surprising chemistry of organometallic (or metalorganic) complexes means that atomic-scale simulations are an important way to elucidate structure, bonding, and reactivity. In this talk, we look at simulations of organometallic complexes as precursor molecules for the deposition or etching of materials. Chemical vapor deposition, vapor phase epitaxy, atomic layer deposition, and atomic layer etching comprise a family of materials processing techniques that all depend on three key precursor properties: volatility, reactivity, and decomposition, and these are the properties focused on in this talk. For example, precursors for atomic layer deposition should ideally show a wide temperature range for reactivity, between a low evaporation/sublimation temperature and a high decomposition temperature. However, computing physical properties (such as volatility) and chemical reactivity (such as ligand exchange or adsorption to surfaces) is also important for organometallics in homogeneous and heterogeneous catalysis. For reactivity, we present simulation workflows integrating quantum mechanics with automatic structure generation, so as to investigate the thermodynamics of adsorption during growth or etching and the bond dissociation reactions that are typical of thermal decomposition. Both approaches are based on density functional theory calculations with special efficiency settings for metal complexes. In addition, we present a new machine-learnt model that predicts evaporation/sublimation temperature as a function of vapor pressure for organometallic complexes of the fifty most common metals and semi-metals to an average accuracy of +-9 degrees C. The three key properties can thus be quantified in silico, either for in-depth study of a single system or for high-throughput computational screening of candidate molecules.
The new solution to the induced fit docking problem: How IFD-MD rapidly and reliably predicts accurate ligand binding
Scientists from Schrödinger’s Structure Prediction, Structure Refinement, and Drug Discovery Groups recently published Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein-Ligand Binding in The Journal of Chemical Theory and Computation. The authors found that Schrödinger’s IFD-MD is able to obtain an accurate structure to use in structure-based drug design (SBDD) more quickly and easily than experimental determination.
Dr. Ed Miller, Product Manager Prime Structure Prediction Group, describes our next generation induced fit docking workflow which provides an accurate and reliable way to determine binding modes of novel ligand scaffolds.
Explain what this paper means. Why is it so exciting?
It’s fairly common in drug discovery for researchers to have access to a structure of their target of interest, or of a close homolog. And yet despite these pre-existing structures, a single side chain, a few atoms even, can render that structure useless for your particular drug discovery project. Obtaining an accurate characterization of the protein-ligand binding mode for your particular compound series is a key starting point for rigorous structure-based modeling efforts, such as free energy methods, to accurately predict affinity. Structures in complex with other ligand scaffolds, not of interest to the drug discovery program, have limitations for scientists designing novel scaffolds.
Historically, significant time and money has been spent to overcome this challenge by obtaining new experimental x-ray crystal structures bound to a team’s own small molecule. This can cause project timelines to extend and delay. For some proteins such as membrane proteins or GPCR’s, obtaining an experimental structure is extremely difficult, if not impossible. There has been a real need for computational methods that can solve this issue in a fraction of the time.
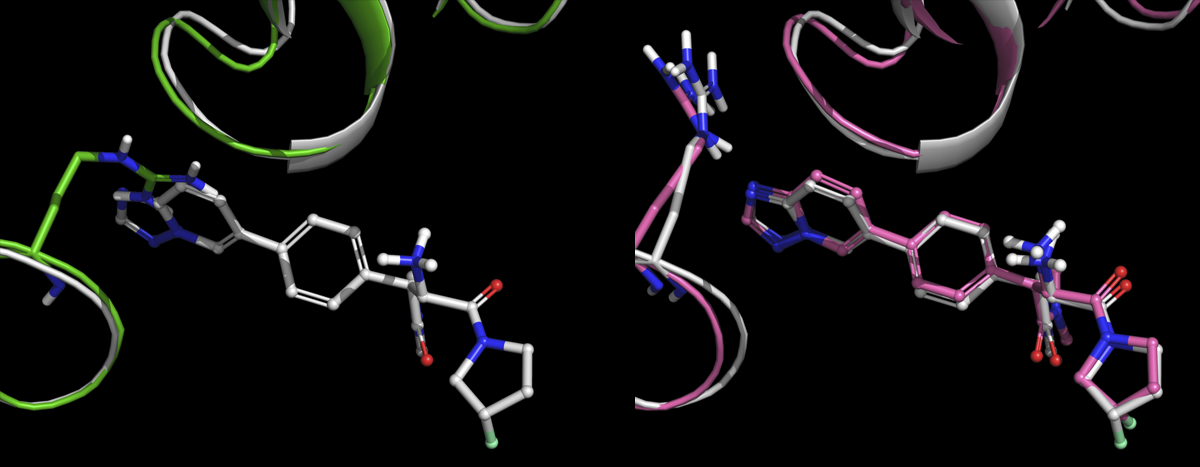
What this paper shows is that by using a new technology called IFD-MD we can produce very highly quality structures and predict protein-ligand binding modes reliably and efficiently for these exact types of problems.
However, for a method like IFD-MD to impact projects, people have to trust the computational models as much as they trust an experimentally-derived crystal structure. They need to have confidence they can use the model prospectively to guide predictions, even in the absence of an experimental structure. That’s a tall ask, but what’s exciting is by challenging and validating the models upfront with free energy calculations (FEP+), scientists can confidently use structures produced by IFD-MD to impact active projects. For example, if FEP+ results using the IFD-MD produced structural model show strong correlation retrospectively, the scientist can have confidence in using the model prospectively. And indeed in the paper, we show this to be the case, prospectively, for two separate internal programs.
What is the difference between Schrödinger’s IFD software (Induced Fit Docking) and IFD-MD?
More than a decade ago, Schrödinger introduced an automated induced fit docking (IFD) algorithm, based on combining our Glide docking program with our Prime protein modeling program. The Glide/Prime IFD methodology has been used extensively in drug discovery projects in the pharmaceutical and biotechnology industries up to the present day. However, limited sampling sometimes means IFD fails to identify or accurately rank native-like protein-ligand binding poses.
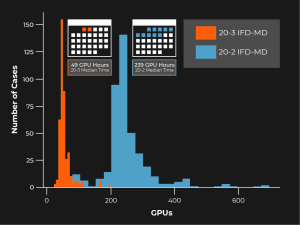
Five years ago, Schrödinger embarked on a project aimed at improving the accuracy and reliability of IFD. The result of these efforts is IFD-MD (Induced-Fit Docking Molecular Dynamics). The method is computationally much more efficient than brute-force molecular dynamics (MD) simulations, and can easily be completed overnight using modest cloud computing resources. In fact, we’ve even made some significant improvements in speed in recent product releases (Figure 1).
Figure 1: Over a set of 413 cross-docks the median compute costs drops from 240 GPU hours per calculation to 49 GPU hours per calculation
We also found that a very high fraction (90% or better) of both training set and test cases reproduce key features of the crystal structure, and yield molecular dynamics trajectories that are very similar to those obtained when starting from the crystal structure.
We subsequently tested the performance of IFD-MD for five proprietary systems investigated in the course of Schrödinger’s in-house drug discovery efforts and asked the question:
“Could one have predicted a proprietary structure with an accurate protein-ligand binding pose using only the publically available structures that were present at the time the drug discovery project was initiated?”
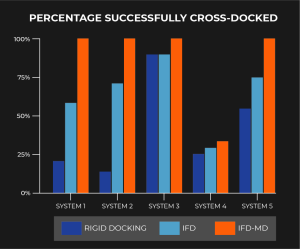
The results of our cross-docking experiments (i.e. docking with a different small molecule than the original experimentally-bound small molecule) are described in the chart below (Figure 2). We compared results across rigid receptor docking method (GlideSP), our original IFD method, and our new IFD-MD method.
Figure 2: performance of IFD-MD for five proprietary systems investigated in the course of Schrödinger’s in-house drug discovery efforts
What was unique about this validation is that protein-ligand systems were not eliminated based on presence or absence of backbone motion or if the template ligand fully occupied the binding site of the target molecule. In fact System 4, the only system that did not consistently produce a successful cross-docked complex 100% of the time, does require significant backbone reorganization which is currently beyond the scope of this method. However, even in prospective cases where severe backbone motion may be unknown but present, testing and validating IFD-MD models using FEP+ de-risks this limitation. Most excitingly, IFD-MD outperforms in all five systems compared to GlideSP and IFD.
Can you describe the IFD-MD workflow?
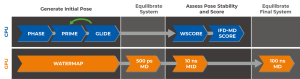
Years of development concluded in the IFD-MD workflow summarized in Figure 3. IFD-MD integrates multiple Schrödinger tools into a single solution for predicting binding poses, and operates as a series of coupled CPU and GPU jobs.
Figure 3: IFD-MD workflow
Initial pose generation is done using pharmacophore docking (Phase) followed by structure refinement using Prime and redocked with Glide in an iterative process. Hydration sites are estimated by calculating their thermodynamics properties (WaterMap) and used for water placement in the binding pocket. After a short system equilibration the stability of the ligand poses are assessed using metadynamics (MtD) simulations and scored.
You note that the IFD-MD method is computationally much more efficient than brute-force MD simulations. Can you explain that?
Perhaps the most straightforward approach to induced-fit docking is to simply simulate, with MD, the ligand traveling from solution into the binding site, with all the atoms in the system, the receptor, the ligand, the solvent, free to move. This is the brute-force approach. There are groups which have shown that it can be done, D.E. Shaw Research most famously. But to do this reliably is very time consuming and expensive, and without a large number of simulations across a range of systems, it’s not clear precisely what is necessary and sufficient to get reliable results.
In an active project, the uncertainties one has to consider start to escalate. There’s alternative tautomeric states, stereoisomers, alternative ligand series, and lots of other variables. Brute force MD simulations are just too expensive to be practical here because each of these variables require a separate simulation, not to mention the valuable time spent by the scientist in having to analyze the large volumes of data generated by brute force MD.
Absolutely. In that publication, the authors state that the main reason limiting the use of FEP+ in some of their discovery projects is a lack of structural data. In particular, protein conformational changes, unresolved atoms in an existing crystal structure, or uncertainty in the binding exist as possible challenges. The induced fit docking algorithm we present in this publication is intended to significantly improve these issues in active drug discovery projects.
Any final thoughts?
Endemic in many computational methods is the rigid-receptor approximation. This approximation has historically shown great utility, but by stepping past it with induced fit methods, a whole realm of new, more accurate simulations become possible. In the near future, we’ll be pushing the limits further of how IFD-MD can impact active drug discovery projects.
Author
Edward Miller
Senior Director of Protein Structure Modeling
Edward Miller joined Schrödinger in 2014, and is responsible for advancing the domain of applicability of structure-based drug discovery into challenging targets and off-targets. Dr. Miller obtained his PhD from Columbia University, where he was awarded a DOE research fellowship. His thesis work with Professor Richard Friesner involved developing methods to accurately model loop conformations across a broad array of protein families. His recent work has been focused on methods development for induced fit docking and protein structure refinement.