Peter O. Stahl Advanced Design Forum 2026
- May 14th-15th, 2026
- Wayzata, Minnesota
Schrödinger is excited to be participating in the Peter O. Stahl Advanced Design Forum taking place on May 14th – 15th in Wayzata, Minnesota. Join us for a presentation by Anand Chandrasekaran, Product Manager, Materials Science Informatics at Schrödinger, titled “Generative AI for Materials.”
Empowering the Digital Chemistry Laboratory: Generative AI, Agentic Workflows, and the Future of Materials Design
Speaker:
Anand Chandrasekaran, Product Manager, Materials Science Informatics at Schrödinger
Abstract:
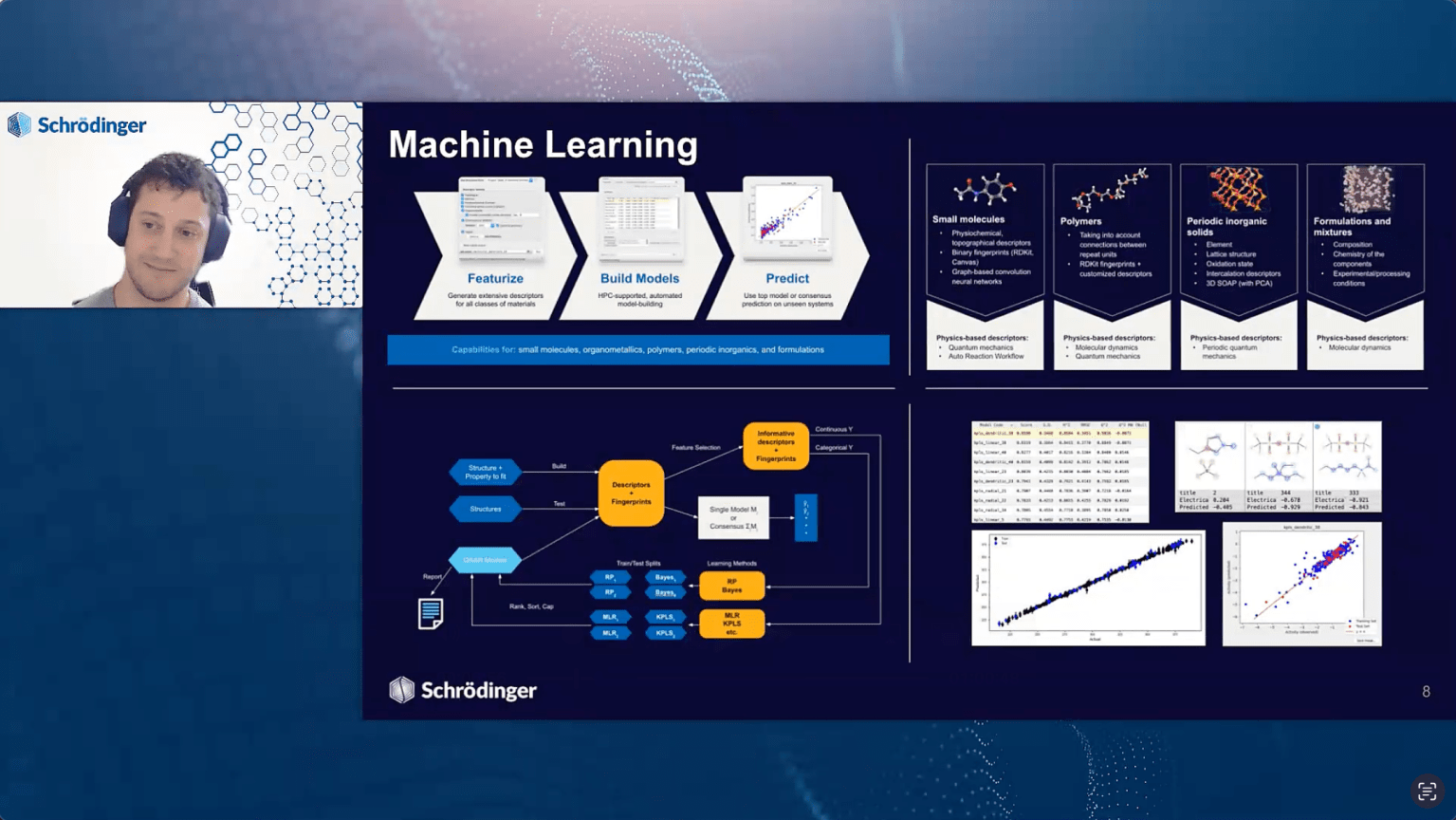
The rapid evolution of artificial intelligence is shifting materials science from expensive, trial-and-error experimentation to autonomous, data-driven design. Drawing on recent advancements at Schrödinger, this talk will address the efficient adoption of AI, its impact on daily scientific practice, and strategies for future-proofing R&D organizations. First, we will explore how AI accelerates existing operations and enables new opportunities. By leveraging Machine Learning Force Fields (MLFFs) like MPNICE, we can drastically speed up computationally expensive quantum mechanics and molecular dynamics workflows while maintaining ab initio accuracy. Furthermore, Generative AI capabilities like REINVENT unlock the true de novo inverse design of novel molecules and complex formulations optimized for specific target properties using reinforcement learning. Next, we will examine how AI enhances autonomy in materials design. We will highlight Formulation Machine Learning and Optimization, which maps chemical structure and composition directly to physical properties to autonomously suggest the “next best experiment”. We will also introduce agentic AI, such as Schrödinger’s digital chemistry assistant, demonstrating how expert digital assistants capable of multi-tasking, pipelining jobs, and executing complex research projects are fundamentally altering how scientists interact with computational platforms. Finally, we will discuss how a digital chemistry strategy built on thesynergy of physics-based modeling and machine learning is the most effective way to future-proof AI adoption. By utilizing rigorous physics-based simulations to generate massive, highly accurate training datasets, organizations can overcome data scarcity limitations and confidently extrapolate into the vast space of synthesizable chemistry.










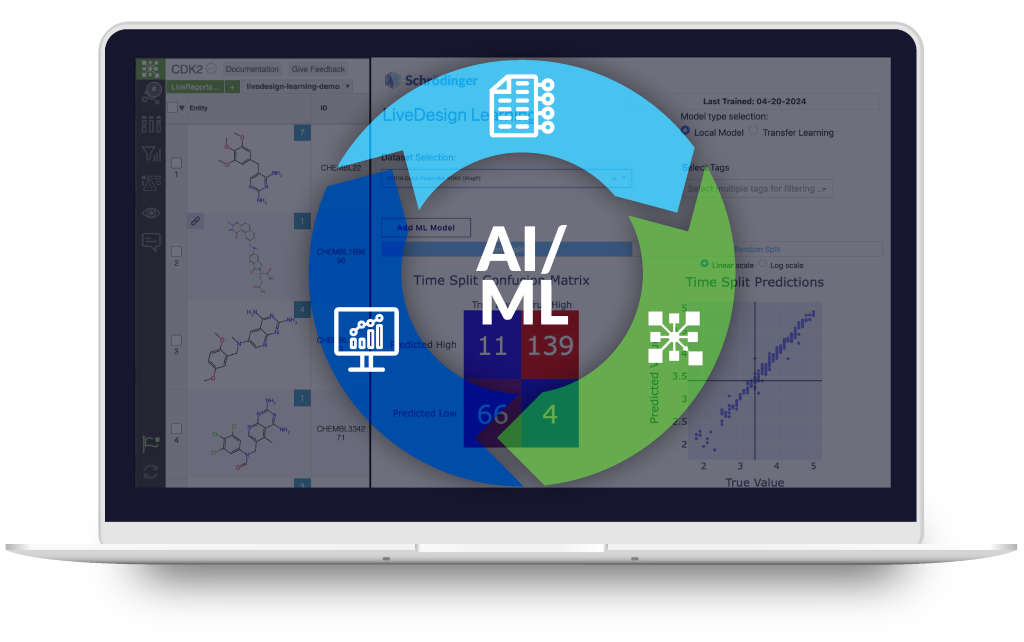















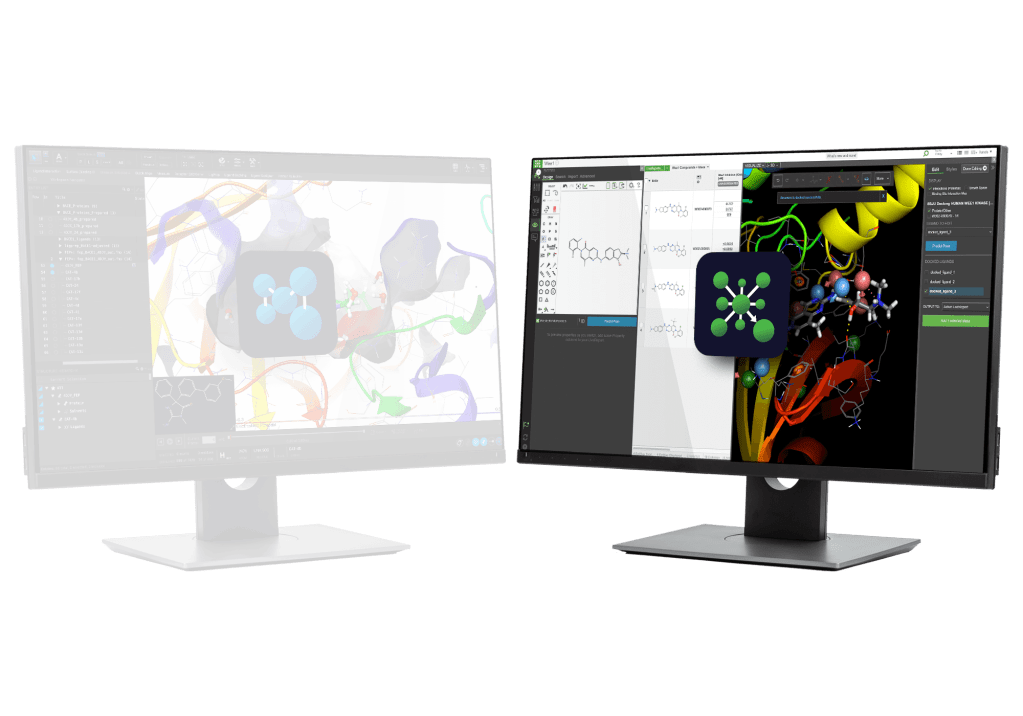
 AI-assisted, intuitive graphical interface to model and interpret molecular interactions
AI-assisted, intuitive graphical interface to model and interpret molecular interactions