Computationally-Guided Drug Formulation Webinar Series
- September 11th, 2024 – May 14th, 2025
- Virtual
A smart, strategic drug formulation can efficiently advance your drug development projects and inform downstream processes. Advances in molecular modeling and machine learning are enabling atomistic-level insights and the ability to evaluate large numbers of candidate materials and formulations prior to experiments.
Computationally-Guided Drug Formulation Webinar Series – Seven webinars in which we demonstrate how the latest computational modeling tools are impacting the various steps in the pharmaceutical formulation process. In each webinar we feature an expert from Schrödinger sharing valuable insights and practical applications on a key topic. Watch the recordings to learn how to optimize your formulation process with structure-based insights and efficient parameter screening.
- May 14, 2025
Computational insights into polymer excipient selection for amorphous solid dispersions
Speaker: Andrea Browning, Senior Director for Polymers
Watch now - April 8, 2025
Accelerating pharmaceutical formulations using machine learning approaches
Speaker: Anand Chandrasekaran, Senior Principal Scientist
Watch now - November 6, 2024
Modeling lipid nanoparticles: Self-assembly and apparent pKa calculation
Speaker: John Shelley, Fellow
Watch now - October 23, 2024
Crystal structure prediction workflow for small molecule drug formulation
Speaker: Lingle Wang, Sr. Vice President, Scientific Development
Watch now - October 9, 2024
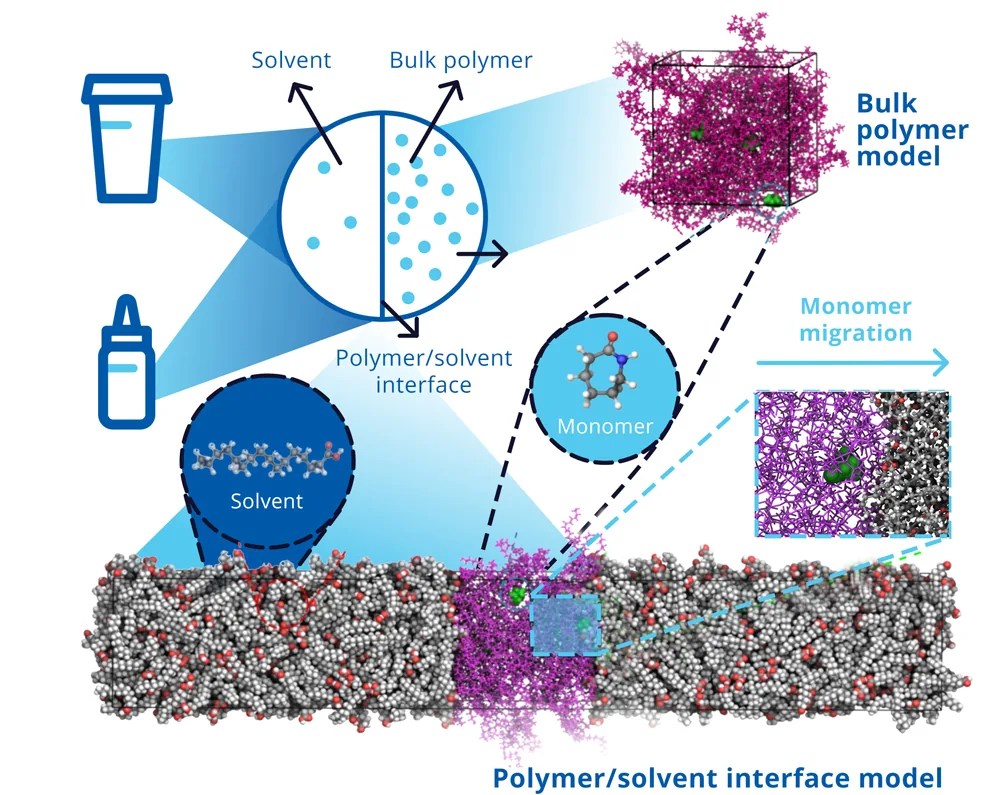
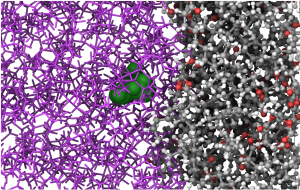
Molecular-level insight into solubility-enhancement via cosolvents and amorphous solid dispersions
Speaker: Ben Coscia, Principal Scientist
Watch now - September 25, 2024
Computational reactivity and catalysis for drug synthesis
Speaker: Michael Rauch, Associate Director, Materials Science
Watch now - September 11, 2024
Characterizing small drug-like molecules with automated computational spectra prediction
Speaker: Art Bochevarov, Research Leader
Watch now