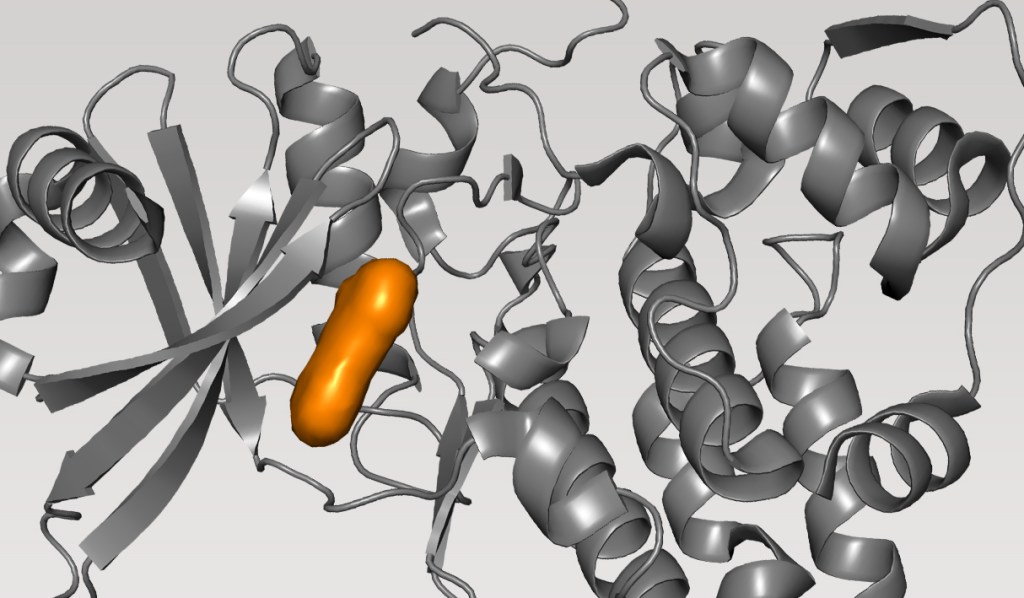
Design of a Highly Selective, Allosteric, Picomolar TYK2 Inhibitor in Clinical Development
Share
Speakers
Craig E Masse
Former Head of Medicinal Chemistry at Nimbus Therapeutics
SVP, Discovery Research at Ajax Therapeutics
Sayan Mondal
Former Senior Director at Schrödinger Therapeutics Group
Abstract
In this webinar, we will highlight key moments from the discovery of this potentially best-in-class selective, allosteric, picomolar inhibitor of TYK2. This case study demonstrates various innovations in modeling to meet challenges over the course of the program, targeting both orthosteric and allosteric binding sites, including:
The first application of large-scale free energy perturbation (FEP+) in drug discovery
Extensive use of a novel physics-based method to predict solubility
The first discovery of novel picomolar cores with FEP+
As these computationally-guided, structure-based discovery strategies are now increasingly deployed across industrial drug discovery projects, this program represents one of the first examples of the quality of the resulting end molecule as it moves through clinical trials.
Bifunctional molecules with two ligands and a linker are able to recruit an E3 ligase to the target protein, leading to the degradation of the target protein. The design of bifunctional degraders is facing a number of challenges; 1) optimization of the warhead ligands and the linker attachment points to avoid disruption of on target binding; 2) given the complex structure, optimization of the binding potency of the degraders; 3) prediction of ternary structure models. The first challenge is essentially the same as small molecule design which is well suited for FEP+. In this Webinar we will show how physics-based simulations such as enhanced sampling and FEP can be applied to the second and third challenges.
Dramatically improving hit rates with a modern virtual screening workflow
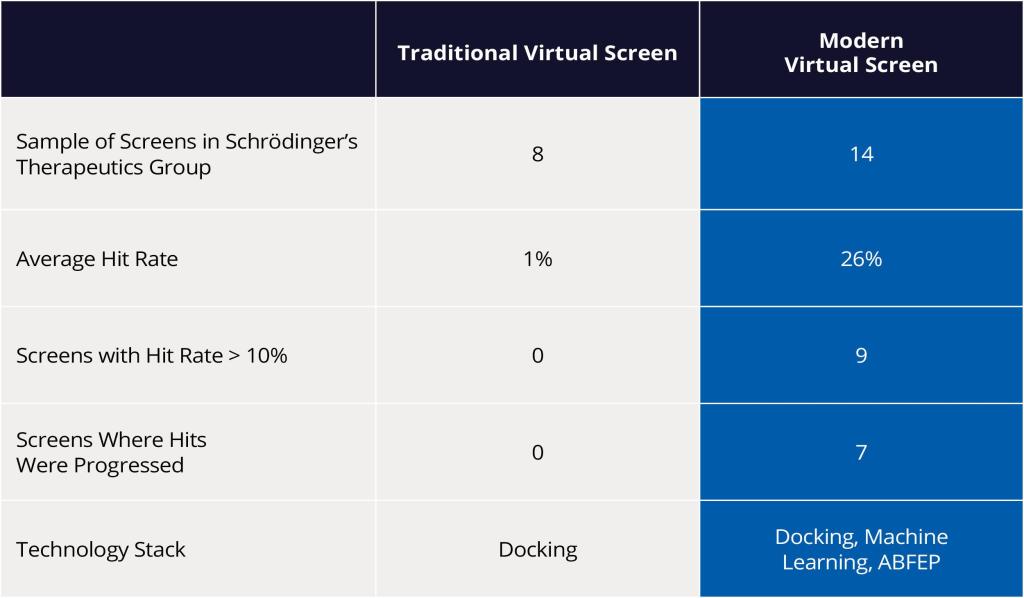
Scientists from Schrödinger’s Therapeutics Group leveraged a modern virtual screening workflow powered by ultra-large scale docking and absolute binding free energy calculations to achieve a double-digit hit rate for diverse protein targets.
Schrödinger developed a modern virtual screening workflow for small molecule ligands and fragments that enabled Schrödinger’s Therapeutics Group to repeatedly achieve unprecedented success in its hit discovery efforts
The modern virtual screening workflow efficiently screens ultralarge libraries of up to several billion purchasable compounds with unrivaled accuracy, through machine learning enhanced docking and absolute binding free energy calculation technologies
The workflow was successfully applied to a broad range of targets across multiple screening campaigns, for both whole ligands and fragments
Schrödinger’s Therapeutics Group used the workflow to identify multiple experimentally confirmed hits with diverse chemotypes while dramatically narrowing down the number of compounds made or purchased and assayed in the lab — frequently achieving double-digit hit rates
Background
For years, hit discovery efforts using traditional virtual screening (VS) approaches have suffered from low hit rates, typically 1-2% in Schrödinger’s experience, which means that 100 compounds would have to be synthesized and assayed for 1-2 hits to be identified. These challenges have largely been attributed to two key factors:
First, traditional VS campaigns have been limited to libraries in the hundreds of thousands to a few million in size, providing limited coverage of chemical space. This is particularly critical for difficult-to-drug targets, where the random hit rate in the library is expected to be low, hence fewer hits are expected to be recovered with smaller libraries. In recent years, the emergence of ultra-large commercial chemical libraries such as Enamine REAL and research demonstrating the value of screening large libraries has further driven the need for technologies that can efficiently screen ultra-large chemical space.1
Second, traditional VS methods have been limited by the inaccuracy of the scoring methods utilized to rank order different ligands, such as GlideScore. Given a static view of the complex geometry and an approximate treatment of desolvation, such empirical scoring functions aren’t theoretically suited to quantitatively rank compounds by affinity. Thus, while docking is a powerful technology for early enrichment, ligand docking scores are not expected to and generally do not correlate with measured potency.
As a result of these limitations, most resources spent on virtual screens using traditional methods are often wasted. A more cost-effective and efficient approach to accurately screen ultra-large libraries is required to improve the success of virtual screens and ensure it is a viable path for hit discovery.
Design Approach
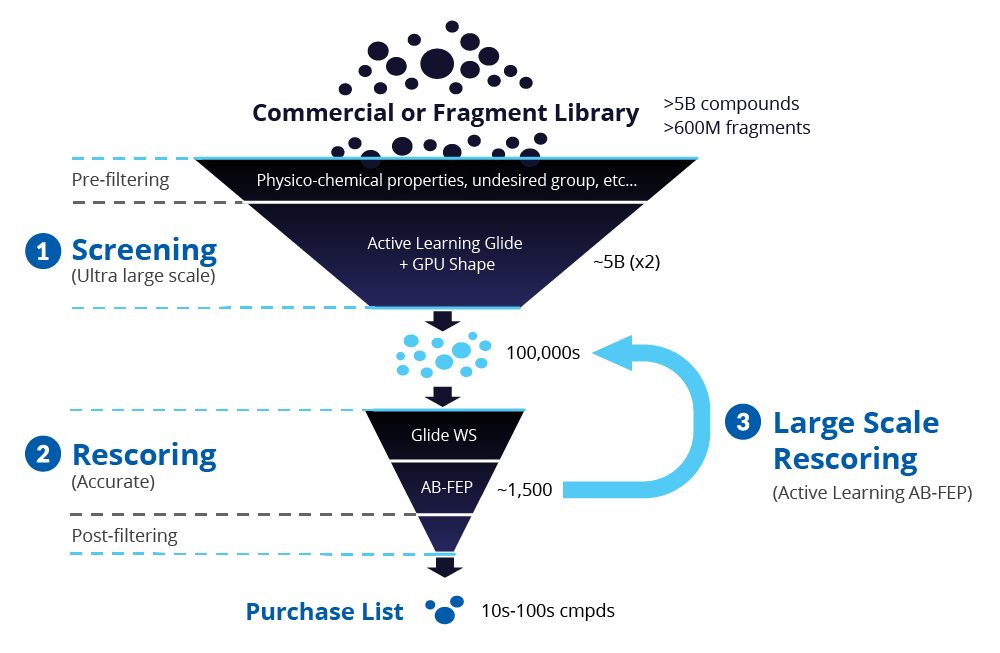
Over several years, Schrödinger’s Therapeutics Group has selected a number of challenging targets with the goal of identifying potent hit molecules. The group turned to a modern VS workflow, leveraging machine learning-guided Glide docking and highly accurate Absolute Binding FEP+ (ABFEP+) calculations, to screen and rescore ultra-large chemical libraries in a way that minimizes wet-lab costs and time while increasing the number and quality of hits available for hit-to-lead progression (Figure 1).
Figure 1: Overview of Schrödinger’s modern virtual screening workflow.
Step 1: Ultra-large scale screening for small molecule libraries
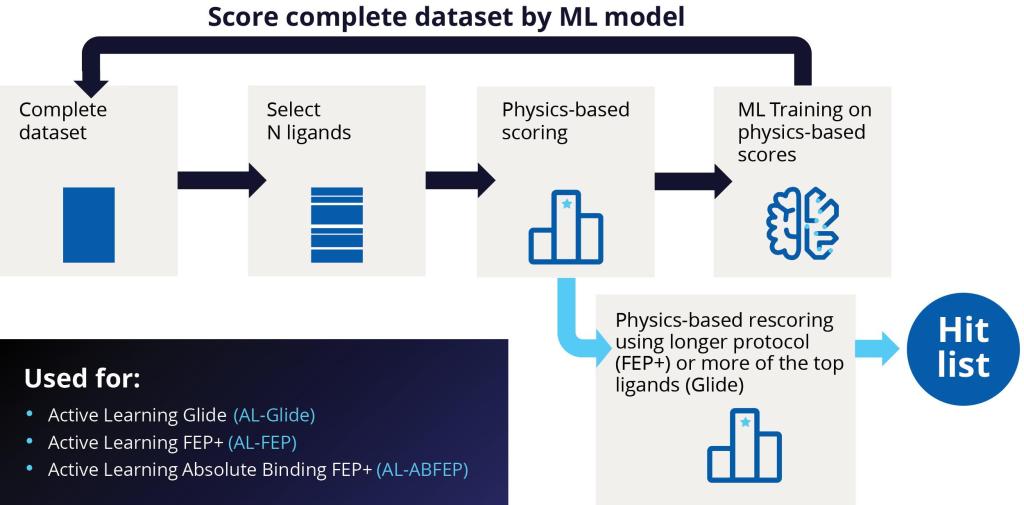
Starting with libraries on the order of several billion compounds (or libraries of up to 500 million for fragments), the team performed prefiltering based on physicochemical properties to eliminate any undesired groups. Next, they carried out a highthroughput virtual screen with Active Learning Glide (AL-Glide), in order to quickly identify the most promising compounds. Active learning is an effective supervised learning strategy that prioritizes training data for the next round of training based on a well-defined objective. AL-Glide combines machine learning (ML) with docking so that enrichment with docking can be applied to libraries of billions of compounds.2 By using this approach, only a fraction of the library is docked, reducing the computational cost significantly to a more reasonable level.
At the start of an active learning cycle, a manageable batch of compounds is selected from the complete data set of library compounds and docked. These selected compounds are then taken and added to the training set. The model is then trained on new information and continues to iterate this process as the machine learning model becomes a better and better proxy for the docking method (Figure 2).
This ML-guided docking model can evaluate compounds much more quickly than brute force docking. While the typical docking calculation with Glide might take an average of a few seconds per compound, the ML model can evaluate or make a prediction significantly faster, leading to a drastic increase in throughput. As a result, the ML-generated model is used to evaluate the entire library.
After completion of the AL-Glide screen, the team performed a full docking calculation using Glide on the best scored compounds, typically in the range of 10-100 million compounds.
Figure 2: Schematic showing an active learning workflow for hit discovery.
Step 2: Rescoring
The most promising compounds based on Glide docking scores were then selected and subjected to a rescoring step using Glide WS, a sophisticated docking program that leverages explicit water information in the binding site to enrich active molecules over Glide alone, in large part due to improved pose prediction. Glide WS helps identify better compounds to pass to ABFEP+, the next scoring step, and to provide more reliable binding poses, reducing false positives in the process.3
Compounds with the best enrichment scores from docking are selected for rigorous rescoring with ABFEP+. ABFEP+ is a protocol in Schrödinger’s FEP+ technology that allows the accurate calculation of binding free energies between the bound and unbound states of the ligand/protein complex (Figure 3).4,5 ABFEP+ has proven to reliably correlate with experimentally measured binding affinities. Unlike relative binding FEP+, ABFEP+ does not require a similar, experimentally measured reference compound as a starting point. Because ABFEP+ can evaluate and accurately score diverse chemotypes, it is a linchpin technology to discover the most potent compounds in a virtual screen campaign.
Step 3: Large-Scale Rescoring
ABFEP+ is computationally expensive when compared to Relative Binding FEP+ (RB-FEP+), requiring multiple GPUs per ligand and approximately 4x more compute time. It is generally only practical to run thousands of ABFEP+ calculations on a hit discovery campaign. In order to realize the true enrichment benefit of ABFEP+, an active learning approach is utilized to score a much larger number of compounds.
Figure 3: Thermodynamic cycle for Absolute Binding FEP+ (ABFEP+). As part of the modern virtual screening workflow on discovery programs, the accuracy provided by the rigorous calculations in ABFEP+ consistently showed very early enrichment in top actives and reduced false positives, which is critical when screening ultra-large libraries. * Compounds with molecular masses between 100 and 250 Da
Applying the modern virtual screening workflow to fragments
Experimental fragment screening has led to multiple FDA-approved drugs and clinical candidates. By adapting this modern workflow to fragment screening, Schrödinger’s Therapeutics Group has successfully scaled up screening to millions of fragments, as compared to 3k to 30k fragments screened by traditional HTS.
The in silico approach addresses a fundamental limitation of experimental fragment screening — the fragments need to be soluble enough to be assayed at high concentrations (100 μM to mM) against various targets. However, estimating the potency rigorously in silico is not limited by solubility, so the potency of the fragments can be assessed and subsequently pursued if they are predicted to be soluble enough given their estimated potency. The binding potency to the specific target is computed using active learning ABFEP+. Priority fragments are finally evaluated for solubility in silico at predicted potency using Solubility FEP+.6 In essence, the approach achieves scale by inverting the problem of potency and solubility, enabling the discovery of highly potent and ligand efficient fragments that would not exist in experimental fragment libraries.
To date, the team has completed a total of nine large fragment-based* virtual screens on multiple challenging targets, including one with a homology model. All nine screens yielded multiple potent ligand efficient hits, ranging from low nM to 30 μM in potency and double-digit hit rates.
Impact of Schrödinger’s modern VS workflow on hit rates across multiple projects and targets
Using these modern VS approaches, scientists at Schrödinger were able to demonstrate a drastic improvement in hit rates compared to traditional screens. As a result, several diverse hit compounds with high predicted binding affinity were identified, acquired, experimentally tested and confirmed as hits — resulting in an impressive doubledigit percentage hit rate (Figure 4).
Figure 4: Impact of transitioning from traditional to modern virtual screening on hit rate.
Conclusion
In silico hit identification has long relied on smaller scale libraries and lower accuracy methods to screen the chemical space, resulting in low hit rates and largely wasted wet lab resources.
By transitioning to a modern workflow that leverages rigorous physics-based methods, including absolute binding FEP+ combined with machine learning, Schrödinger’s Therapeutics Group has been able to successfully apply the workflow to a range of diverse targets across several projects and achieve a reproducible doubledigit hit rate. In the process, the team dramatically reduced the number of compounds synthesized and tested to reach the project’s lead candidate, reducing overall costs and project timelines.
This strategy empowers drug discovery teams by enabling them to explore the fast-growing ultra-large chemical libraries. It allows efficient navigation through the vast maze of chemical space, significantly improving the odds of identifying multiple hits with better properties and selectivity. Moreover, it accelerates the drug development process, leading to the faster discovery of higher-quality, novel drug candidates.
Enabling digital technologies to drive discovery programs
FEP+
Digital assay for predicting protein-ligand binding across broad chemical space at an accuracy matching experimental methods.
Ultra-large library docking for discovering new chemotypes. Lyu J, et al.
Nature. 2019 Feb; 566(7743): 224–229.
Efficient exploration of chemical space with docking and deep learning. Yang Y, te al.
J. Chem. Theory Comput. 2021, 17, 11, 7106–7119.
WScore: A flexible and accurate treatment of explicit water molecules in ligand−receptor docking.
Murphy RB, et al. J. Med. Chem. 2016, 59, 4364−4384.
Enhancing hit discovery in virtual screening through accurate calculation of absolute protein-ligand binding free energies.
Chen W, et al. J. Chem. Inf. Model. 2023, 63, 10, 3171–3185.
Accurate calculation of the absolute free energy of binding for drug molecules.
Aldeghi M, et al. Chem. Sci. 2016;7:207–218.
Novel physics-based ensemble modeling approach that utilizes 3D molecular conformation and packing to access aqueous thermodynamic solubility: A case study of orally available bromodomain and extraterminal domain inhibitor lead optimization series.
Hong RS, et al. J. Chem. Inf. Model. 2021, 61, 3, 1412–1426.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
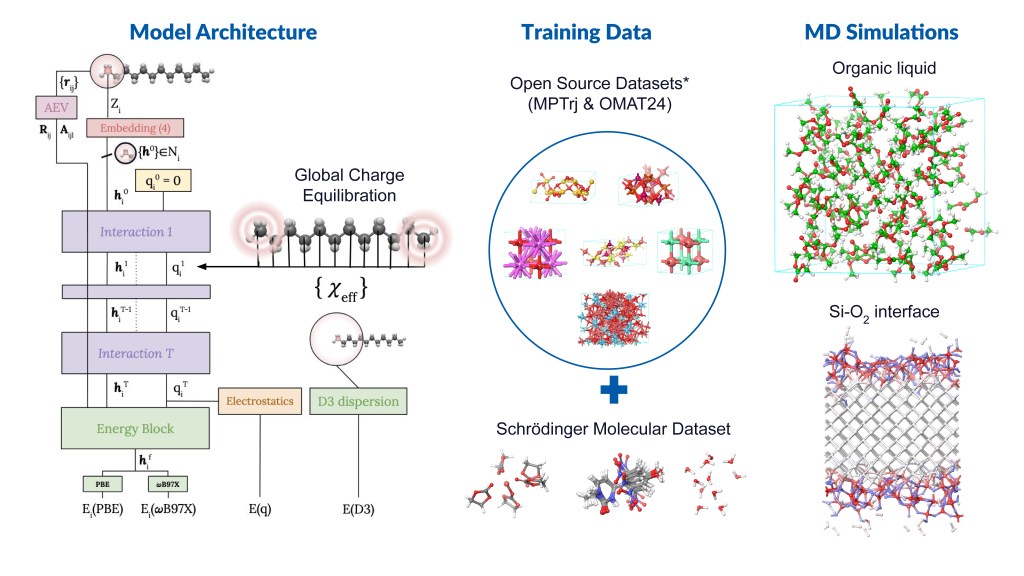
Machine learning force fields (MLFFs), also known as machine learning interatomic potentials, represent an intermediate between classical force fields and density functional theory (DFT), maintaining the linear scaling of the former while approaching the accuracy of the latter. Beyond the balance of accuracy and efficiency/cost, MLFFs are enabling new scientific insights by making large-scale and longtime scale simulations feasible for reactive systems. This opens the door to modeling complex materials systems that were previously computationally prohibitive with traditional quantum methods.
Message Passing Network with Iterative Charge Equilibration (MPNICE) is an MLFF architecture developed by Schrödinger for which multiple pretrained models spanning 89 elements are available, and which explicitly incorporates equilibrated atomic charges and long range electrostatics.1 This technological advancement has removed the drawback of previous MLFFs that were limited by the number of unique atomic elements they could model. Furthermore, inclusion of atomic charges and electrostatics through charge equilibration has enabled representation of multiple charge states, ionic systems, and electronic response properties, while simultaneously improving accuracy. In addition to MPNICE, the Schrӧdinger suite also allows users to utilize the Universal Models for Atoms (UMA),2 developed at Meta. This suite of models offers very high accuracy, includes a model that yields good performance for reaction barrier heights for finite systems, and covers the majority of the periodic table.
By integrating state-of-the-art MLFF methods with high-performance OPLS4 or OPLS5 force fields, as well as advanced DFT and molecular dynamics (MD) engines, Schrödinger offers a uniquely powerful platform for materials simulation — positioning us as the leading partner in advanced MLFF technologies. In this application note, we present case studies from materials-intensive industries, including batteries and catalysis.
Benefits of MLFF
Near DFT-level accuracy with orders of magnitude reduction in computational time
Option for GPU accelerated molecular dynamics with Desmond
Large chemical space spanning 89 elements
Specialized force fields for organic, inorganic, and hybrid materials
Figure 1: Illustrative examples of Schrödinger’s MLFF workflow and its applications *Datasets references: Nature Machine Intelligence 2023, 5, 1031–1041; arXiv:2410.12771
Diverse applications of MLFF
Batteries:
Calculate bulk and transport properties, such as diffusion, viscosity, and conductivity of liquid electrolytes
Simulate Li-ion diffusion in solid-state electrolytes and cathode coating materials
Model electrolyte reactivity and SEI formation
OLED materials:
Simulate molecular packing and thin-film morphology
Investigate doping, host-guest, and interlayer interactions
Link device properties to the static and dynamic disorder of molecular systems
Facilitate thermomechanical property prediction
Model charge and exciton transport
Crystal structure prediction:
Rank order organic crystal structures
Adsorption on surfaces:
Study reactivity of multiple adsorbates in extended models of complex surfaces
Reactivity in molecules and solid-state:
Investigate reaction pathways and transition states
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
FEP+ is a widely adopted technology for accelerating small molecule drug discovery programs, with application in hit discovery, hit-to-lead, and lead optimization. The accuracy and utility of FEP+ as a computational assay for the prediction of binding energies is validated extensively, with predictions falling within 1.0 kcal/mol of experimental values on average.1,2 With accuracy approaching lab experiment, FEP+ serves as a digital binding affinity assay for both on- and off-targets which enables drug discovery teams to efficiently optimize molecular profiles and pursue novel chemistry with confidence.
Given the computational cost and complexity of running free energy calculations, adoption is largely driven by expert computational chemists who then manually share results with the project team. To amplify the impact of these methods and improve the agency of individual team members, they should be accessible directly to medicinal chemists and be able to be run at a scale comparable with traditional virtual screening methods.
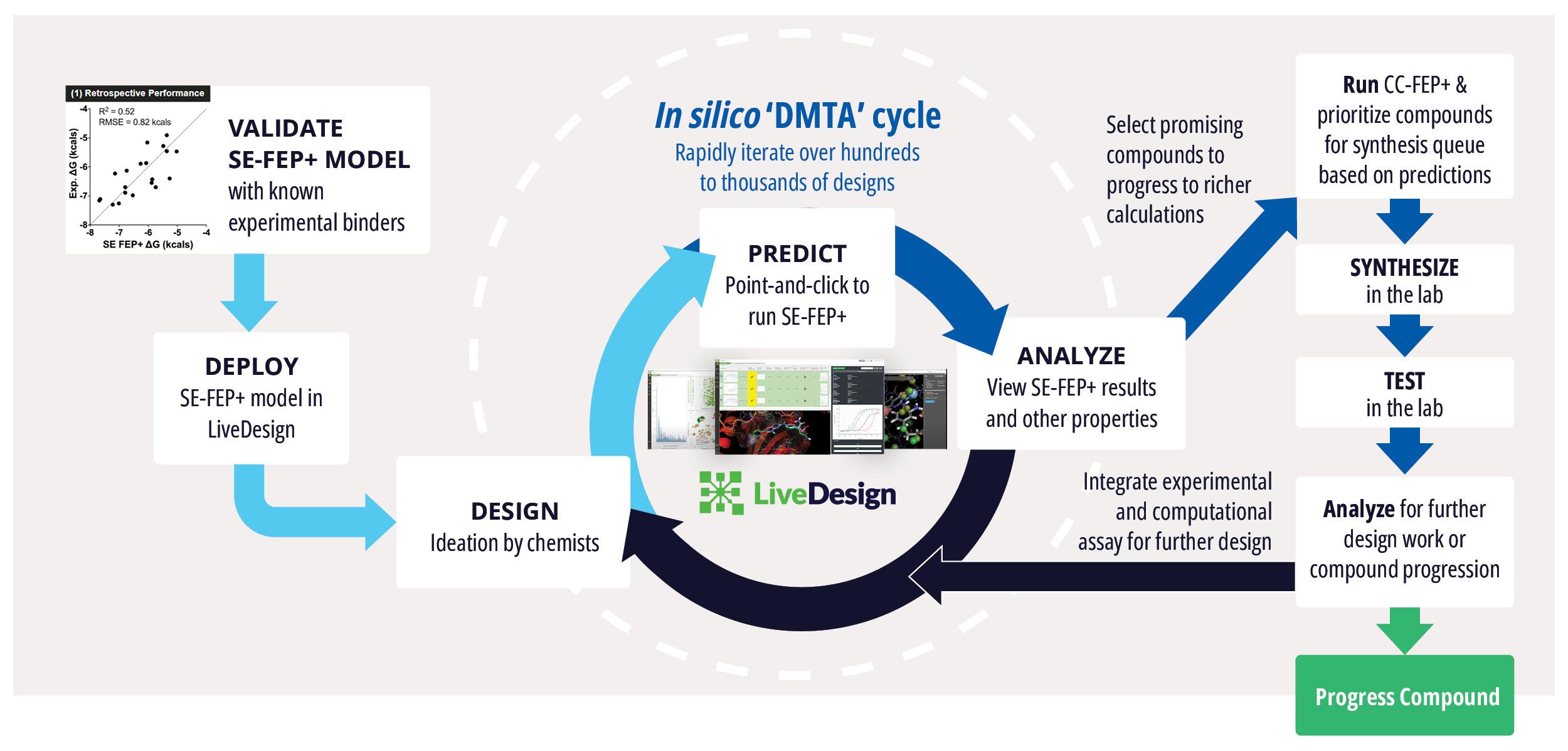
Toward these goals, developments in expanding the accessibility of free energy calculations — in particular single-edge FEP+ (SE-FEP+) — enable a new paradigm in structure-based molecular design. SE-FEP+ simulations can be run in a fraction of the time of full cycle-closure FEP+ (CC-FEP+) simulations and can be conceptualized as ‘push-button’ for execution. This makes it easily deployable in LiveDesign, a cloud-native, collaborative enterprise design platform. This workflow enables all project team members to integrate FEP+ within their design workflows, resulting in a highly interactive and fully in silico design-make-test-analyze (DMTA) cycle where chemists are empowered to digitally test hypotheses and iteratively improve designs prior to compound synthesis (Figure 1).
In one such oncology program within Schrödinger’s Therapeutics Group, the project team was able to improve compound potency over 100-fold by running many iterative in silico DMTA cycles using FEP+ in LiveDesign over the course of a four week period.
Figure 1: Process for deploying and running in silico DMTA Cycles via SE-FEP+ and its potential for tight integration with traditional experimental DMTA cycles.
Single-Edge FEP+:
Accurate prediction of relative binding affinities in a fraction of the time
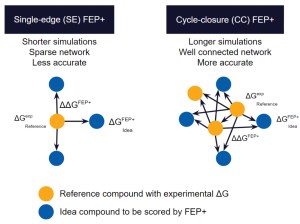
As compared to docking or MM-GBSA calculations, full cycle-closure FEP+ (CC-FEP+) simulations enable highly accurate rank-ordering of compounds, but are compute intensive and complex to set up, reducing their ability to be set up and executed by non-expert molecular modelers.
Single-edge FEP+ (SE-FEP+) simulations have been shown to maintain high predictive accuracy but can often be run ~10x faster than CC-FEP+ simulations in an automated fashion to triage new designs efficiently. Once the SE-FEP+ model is validated, it can be used in a push-button fashion to test new designs that share a common scaffold (i.e., common core or common R-group) with the reference compound and enable efficient scoring of large numbers of idea compounds (Figure 2).
When deployed via a collaborative platform such as LiveDesign, SE-FEP+ gives project teams the ability to run rapid design cycles and prioritize ideas with confidence. The most promising design ideas can then be run through full CC-FEP+ calculations to obtain the most robust potency predictions for informing synthesis queue decisions. If a compound falls outside the criteria established by the computational chemist for running the SE-FEP+ models, that molecule can be added to a queue for running full CC-FEP+ in a more manual setup by a computational chemist.
Figure 2: Schematic representation of Single-Edge FEP+ and Cycle-Closure FEP+. During single-edge FEP+, idea compounds are evaluated against a single reference compound and usually for a fraction of the simulation length compared to full CC-FEP+ calculations, resulting in a faster albeit less accurate calculation. During cycle-closure FEP+, design ideas are usually evaluated against multiple reference compounds in an interconnected map with longer simulation lengths for the highest accuracy results.
A complete digital molecular design lab
LiveDesign is a flexible, cloud-native collaborative working environment for entire discovery teams. The platform democratizes digital design processes and enables more productive design cycles by harnessing the power of medicinal chemistry strategies, advanced cheminformatics, cutting-edge computational chemistry workflows, virtual ‘design and test’ technologies, and centralized access to live project data — all in a single interface.
The platform empowers creativity by democratizing access to powerful predictive modeling workflows, such as push-button free energy calculations, and allows teams to efficiently capture and progress their most promising design ideas. LiveDesign also centralizes team collaboration and decisionmaking — enabling crowdsourcing of design ideas, interactive team-based design, and easy sharing of data with internal and external partners.
Case Study:
Oncology Program SCH-01
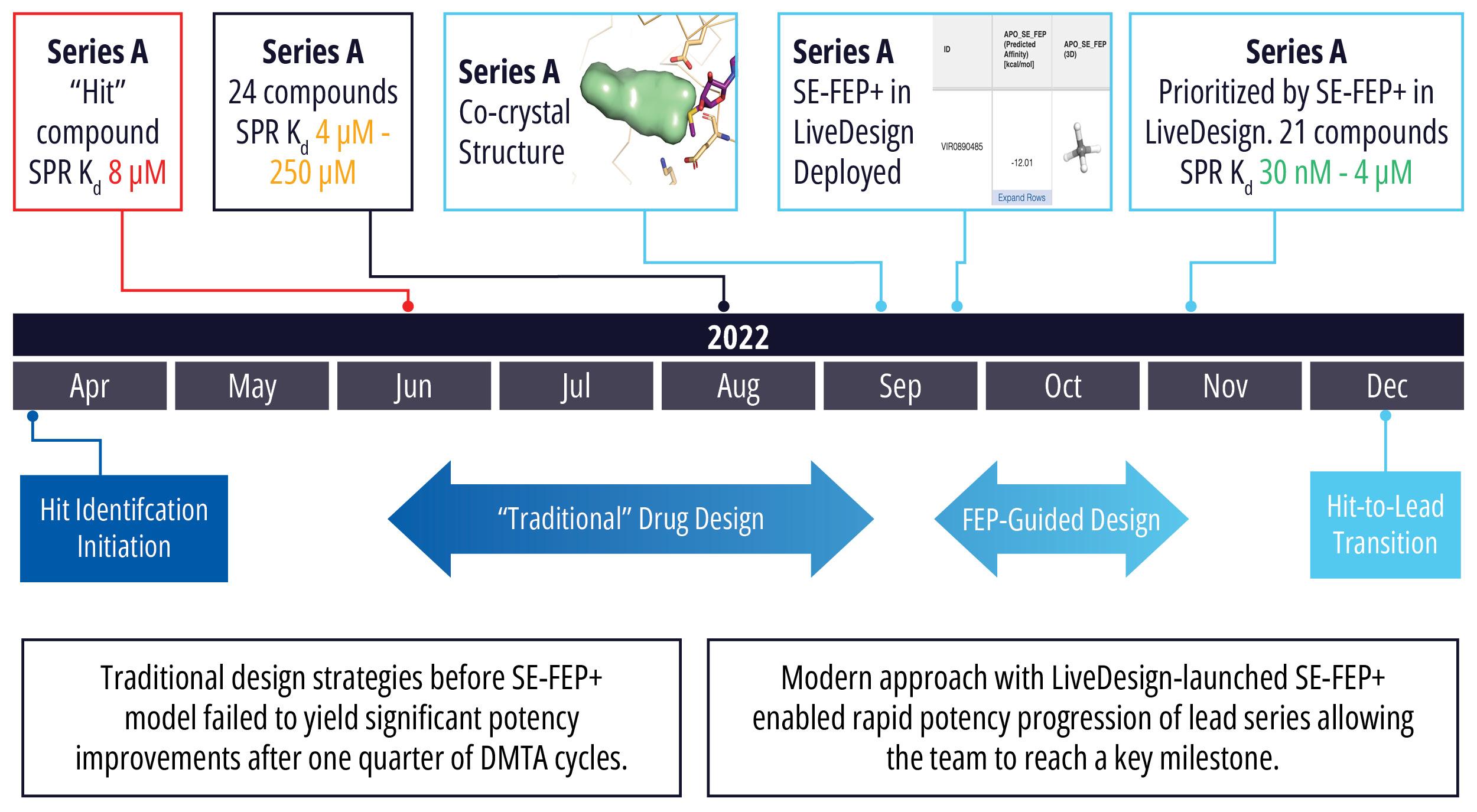
In an early stage oncology drug discovery program driven by Schrödinger’s Therapeutics Group, the project team faced a design challenge to find and optimize an initial hit series. Of the 200 compounds synthesized in the program to date, the most potent molecule had a modest binding affinity of 2.7 μM, which failed to meet the criteria for the hit-to-lead transition. Traditional design strategies had failed to yield significant potency improvements after several months of DMTA cycles, even in the most promising series.
With a new cocrystal structure for this promising series in hand, the team turned to FEP+ to address the potency challenge. An SE-FEP+ model was developed and validated retrospectively using known experimental binding affinities. The SE-FEP+ model was then deployed in LiveDesign, allowing medicinal chemists to design new ideas and independently run SE-FEP+ calculations with the click of a button.
Using this validated model, the project team iteratively designed molecules and digitally assessed their ability to improve potency from the current best Series A potency of 4 μM. During one particular four week period, approximately 400 designs were profiled with medicinal chemistry team members contributing and running SE-FEP+ in LiveDesign for ~65% of the designs. No new compounds were experimentally synthesized and tested in this month, but the team was able to significantly optimize compound potency in silico as predicted by FEP+. Each week, the predicted potency of the designs made by the chemistry team improved until the top scoring compounds were then prioritized for synthesis and experimental profiling (Figure 3).
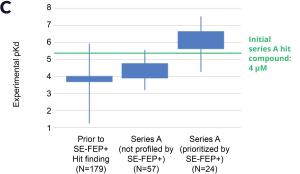
By prioritizing 24 compounds for synthesis based on SE-FEP+ predictions in LiveDesign over this one month period, the project team dramatically enriched the synthesis queue with potent designs. In fact, 21 compounds exhibited improved measured binding affinities of less than 4 μM (Figure 4). Based on this significant progress, the team moved the program forward from the hit identification stage to the hit-tolead stage in a highly accelerated time frame.
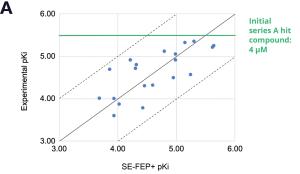
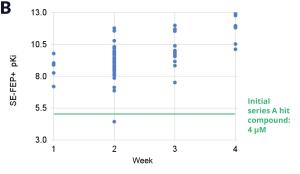
Figure 3: Rapid potency improvements from SE-FEP+ in LiveDesign over a four week period. A) Initial retrospective validation of single edge FEP+ model performance vs experiment (known binders) B) Predicted binding affinity by week for new designs profiled directly in LiveDesign over a four week period demonstrates rapid improvement as the team iterated on idea compounds in silico. C) Confirmed experimental binding demonstrates over 100 fold improvement from compounds prioritized by the SE-FEP+ in LiveDesign workflow compared to those designed prior to SE-FEP+.
Figure 4: Impact of SE-FEP+ in LD on program hit identification timeline
Conclusion:
Deployment of SE-FEP+ in LiveDesign presents a new paradigm for computationally-guided design by democratizing access to powerful, push-button FEP+ calculations to teams of medicinal chemists. It enables chemists to think broadly and creatively when designing ideas by lowering the barrier to in silico testing of ideas before synthesis. Additionally, the workflow enables expert computational chemists to create guardrails around the use of FEP+ by chemistry teams, and focus time and effort on the more complicated full-cycle challenges to the highly triaged compounds.
This workflow can assist in rapid potency progression, optimization of selectivity against known off-targets, and maintenance of potency while optimizing other physicochemical properties. This allows project teams to quickly reach key milestones and design the best possible molecules to meet the target product profile while dramatically reducing the number of compounds synthesized and tested.
How it works
Succcesful deployment of SE-FEP+ in LiveDesign follows these key steps:
A scientist trained in FEP+ creates and validates an FEP+ model on a specific protein-ligand series of interest.
A model execution strategy is determined around permitted structural modifications, licenses, and available compute hardware as these calculations are computationally significant.
The FEP+ model is uploaded into LiveDesign and shared via a user-input constrained model.
In LiveDesign, collaborators design structural modifications to the reference molecule and run SE-FEP+ calculations through the push of a button.
Results are provided directly in LiveDesign, including the corresponding predicted changes in free energy for each design idea, simulation statistics, and the 3D pose of the ligand in the protein.
Teams can continue to ideate and explore modifications to identify promising compounds. The most promising compounds are then progressed and further scored with CC-FEP+ for the highest accuracy binding affinity prediction.
Best Practices
Key points to consider before deploying SE-FEP+ in LiveDesign:
Ensure your SE-FEP+ model is well-validated using known experimental binding affinities.
Educate all project team members on accurate interpretation of model predictions and to build appropriate confidence in the SE FEP+ model. The Schrödinger Online Course, “Free Energy Calculations for Drug Design”, is a useful resource to learn more about FEP+, including a hands-on case study.
Develop template reports in LiveDesign that include automated safeguards around what structural modifications can be run successfully using the SE-FEP+ models.
Ensure sufficient cluster compute resources are secured to run SE–FEP+ simulations efficiently based on anticipated throughput.
Schrödinger is happy to partner with users to share best practices and provide guidance on optimizing FEP+ workflows for discovery programs. Reach out to your local Schrödinger team for assistance.
References
Impacting Drug Discovery Projects with Large-Scale Enumerations, Machine Learning Strategies, and Free-Energy Predictions
Knight et al. ACS Symposium Series, 1397, 2021, 205-226
Advancing Drug Discovery Through Enhanced Free Energy Calculations
Abel et al. Acc. Chem. Res. 2017, 50, 7, 1625–1632
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
compounds scored with rigorous physics-based modeling
226
total compounds synthesized
25months
to discovery of development candidate
Target
CDC7, kinase
Program Type
Schrödinger proprietary program, small molecule
Indication
Hematological cancers and solid tumors
“Through large-scale chemical space exploration with our digital platform, the team was able to overcome several key program challenges, including a potential liability in the lead series. A quick pivot led to the discovery of SGR-2921, which is the most potent CDC7 inhibitor reported to date and possesses strong drug-like characteristics.”
Adam Levinson
Director, Medicinal Chemistry
Schrödinger Therapeutics Group
Design challenge
Cell division cycle 7-related protein kinase (CDC7) is a cell cycle kinase which plays a key role in replication stress response. Inhibition of CDC7 is a long pursued strategy for targeting hematological cancers and solid tumors. While drug discovery efforts focused on the development of small molecule inhibitors of CDC7 span more than 20 years, none have advanced beyond initial Phase I or II studies as most lacked the properties required for successful clinical development.
Given the very high affinity of CDC7 to its natural substrate, ATP, the primary challenge is the development of highly potent inhibitors — ideally picomolar, pM — to be substrate competitive while also balancing selectivity and other drug-like properties. Overall, the goal of the programwas to discover new chemotypes with best-in-class properties.
Better designs through crowdsourced ideation and physics-based modeling
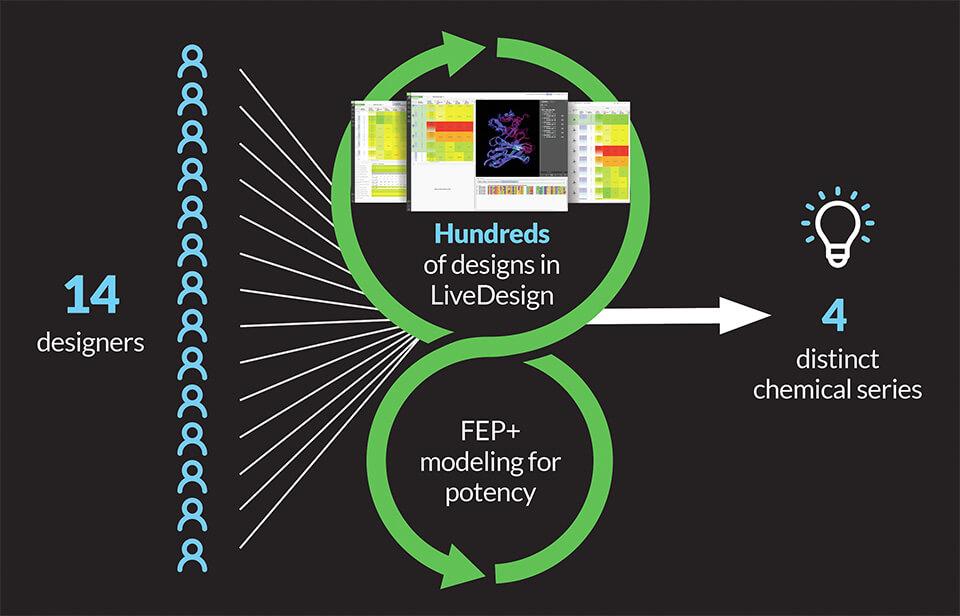
To rapidly iterate through high-quality ideas and analysis, the team crowdsourced ideation amongst 14 designers using LiveDesign, a modeling-enabled collaborative enterprise informatics platform for real-time design. Drawing on a wealth of existing structural and activity data, designs were evaluated through an iterative workflow using relative binding affinities via FEP+, free energy perturbation technology (Figure 1). The accuracy and utility of FEP+ as a computational assay for the prediction of relative binding energies of molecules has been validated extensively, generating predictions within 1.0 kcal/mol of experimental values on average.1 This armed the entire team with a true digital assay, allowing reliable predictions of potency and an unprecedented level of throughput.
Within four months of project start, the team identified four distinct, ligand-efficient chemical series with high affinity, ranging from low nanomolar to picomolar in biochemical assays. These chemotypes served as starting points for further optimization of potency and pharmacokinetic (PK) properties.
Figure 1: Initial iterative crowdsourced ideation using LiveDesign and FEP+ to design novel, potent CDC7 inhibitors.
Overcoming selectivity and PK liabilities with next-generation modeling techniques
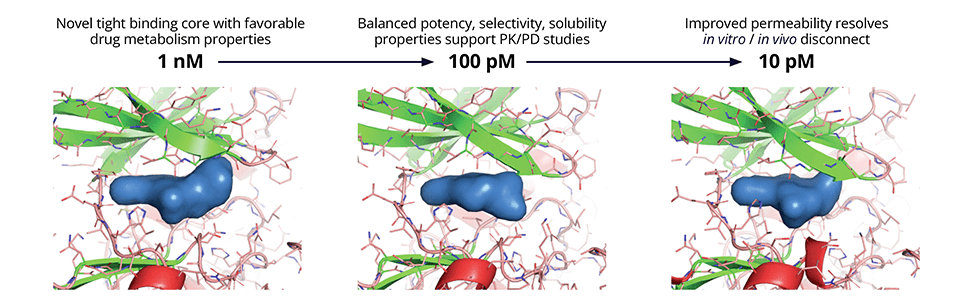
Given the structural similarity inherent within kinase families, achieving sufficient selectivity to mitigate offtarget effects is a core challenge of most kinase drug discovery programs. While the team successfully identified new, attractive chemical starting points, off-target mediated toxicity remained unresolved after the first round of design efforts. To address this selectivity liability, the team created a structural hypothesis for off-target binding using IFD-MD, Schrödinger’s state-of-the-art induced fit docking engine that generates highly accurate ligand binding poses in the absence of experimental structures. Poses resulting from IFD-MD were evaluated by FEP+ to ascertain potency as compared to CDC7. Ultimately, by utilizing this strategy the team identified a proof-of-concept compound with 10,000x selectivity for CDC7 against the off-target in question.
While selectivity was improved, the shift from biochemical potency to cell-based potency was large and variable; this could be attributed largely to suboptimal membrane permeability. A physics-based model for passive membrane permeability (Membrane Permeability) explained the discrepancy observed between experiments and enabled the team to prioritize designs with a consistently lower shift from biochemical to cell potency. Taking into consideration all factors required to satisfy the target product profile (TPP), the team deprioritized the lead series in favor of a new backup scaffold identified by FEP+ that exhibited a superior balance of properties including safety (Figure 2).
Figure 2: Optimization of the lead series using physics-based molecular modeling strategies, in particular to improve kinome selectivity and permeability while maintaining on-target potency.
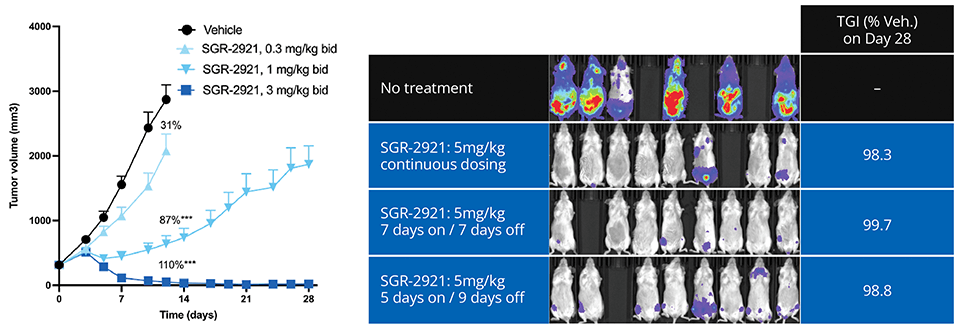
Ultimately, the team’s efforts culminated in the discovery of SGR-2921, with binding potency of ~10 pM in SPR with ligand efficiency > 0.6. SGR-2921 exhibits strong anti-tumor activity in vivo across multiple acute myeloid leukemia (AML) xenograft models, both as a monotherapy and in combination with standard of care agents (Figure 3). Moreover, SGR-2921 demonstrates potent anti-tumor activity in AML patient-derived samples that are resistant to standard of care agent.
Figure 3: (Left) SGR-2921 achieves tumor regression in the TP53 mutated MOLM-16 CDX model; (Right) SGR-2921 demonstrates tumor growth control with intermittent dosing in the MV-4-11 disseminated model.2
Enabling digital technologies to drive discovery programs
FEP+
Digital assay for predicting protein-ligand binding across broad chemical space at an accuracy matching experimental methods.
Advancing Drug Discovery Through Enhanced Free Energy Calculations.
Abel et al. Acc. Chem. Res. 2017, 50, 7, 1625–1632.
Inhibition of CDC7 with SGR-2921 in AML models results in enhanced DNA damage and anti-leukemic activity as monotherapy and in combination with standard of care agents.
Tsvetkov et al. ASH 2022.
Discovery of novel CDC7 inhibitors that disrupt cell cycle dynamics and show anti-proliferative effects in cancer cells.
Tsvetkov et al. AACR 2021.
Huang X, Mondal S, Ghanakota P, Boyles N, Frye L, Gerasyuto A, Greenwood JR, Tang H, Levinson AM. Cyclic Compounds and Methods of Using Same.
U.S. Patent No. WO/2021/113492. 2021.
Mondal S, Tang H,Huang X, Levinson AM, Frye L, Bhat S, Bos PH, Konst Z, Ghanakota P, Greenwood JR. Cyclic Compounds and Methods of Using Same.
U.S. Patent No. WO/2022/197898. 2022.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Stereoisomers of pharmaceutically relevant molecules may have different effects on living organisms. Therefore, knowledge of the absolute stereo-configuration of the synthesized drug is of critical importance. As shown in the FDA guidance, knowledge of the stereochemistry, also referred to as absolute configuration (AC), is required for approved enantiopure therapeutics.1
VCD spectroscopy is a technique that allows one to determine the absolute configuration in solution by comparing experimentally measured and theoretically predicted VCD spectra.2 The VCD spectra of some molecules may show significant variations with the solvent (mainly due to conformational and hydrogenbonding effects). As the choice of the solvent for a VCD measurement may be dictated by the solubility of the molecule, it is important to establish that theoretical VCD computations can account for solvent effects with sufficient accuracy.
R-2-hydroxy-4-phenyl butyric acid ((R)-2H4PBA) is a carboxylic acid whose experimental VCD spectra in chloroform (CHCl3) and dimethyl sulfoxide (DMSO) show significant differences. These differences likely arise from intermolecular interactions taking place in the two solutions. In order to accurately reproduce the experimental spectra by performing VCD calculations, it is necessary to account for these interactions when setting up the calculations.
This white paper demonstrates the combined use of BioTools’ experimental VCD measurements and Schrödinger’s computational VCD predictions for the purpose of not only resolving the absolute configuration of 2H4PBA in two standard solvents, CHCl3 and DMSO, but also demonstrating possible methodology to achieve greater spectral overlap between experimental and theoretical VCD spectra.
We further demonstrate the use of available comparison and alignment algorithms such as the Pearson coefficient3 (PC) found in Schrödinger’s software and the Enantiomer Similarity Index (ESI)/Confidence level found in BioTools’ CompareVOA algorithm.4 Briefly, the Pearson coefficient is a measure of the degree of spectral overlap of the measured and calculated VCD spectra. CompareVOA calculates SNS, the sigma neighborhood similarity in percent, and the ESI, the enantiomeric similarity index (ESI or delta), is the difference between the SNS of the favored enantiomer and the SNS of the opposite (less similar) enantiomer. The larger the ESI, the higher is the confidence level of the visual assignment of AC. Both PC and ESI can be used in tandem for extra assurance.
Experimental spectra and modeling of theoretical spectra in chloroform and DMSO
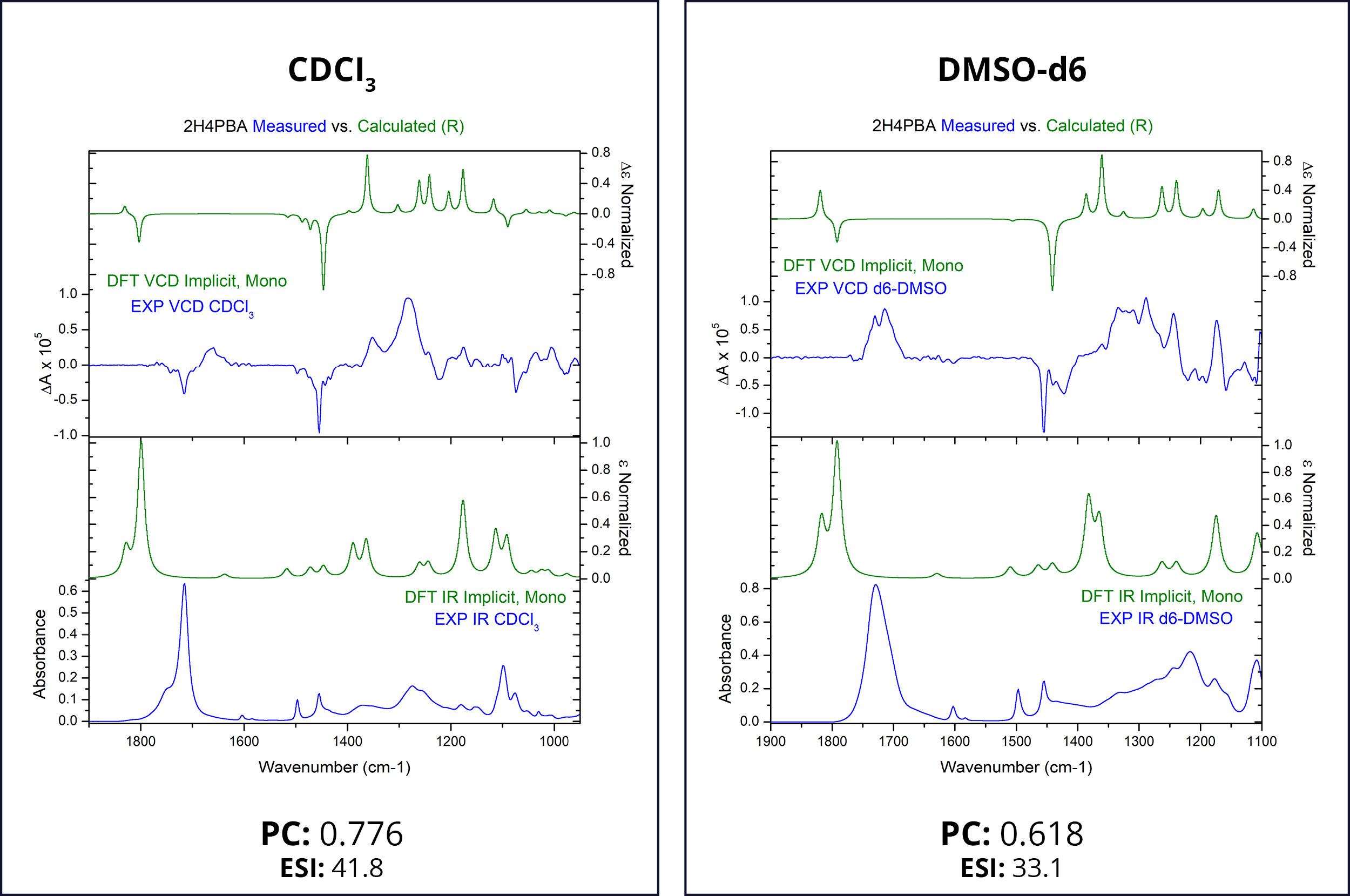
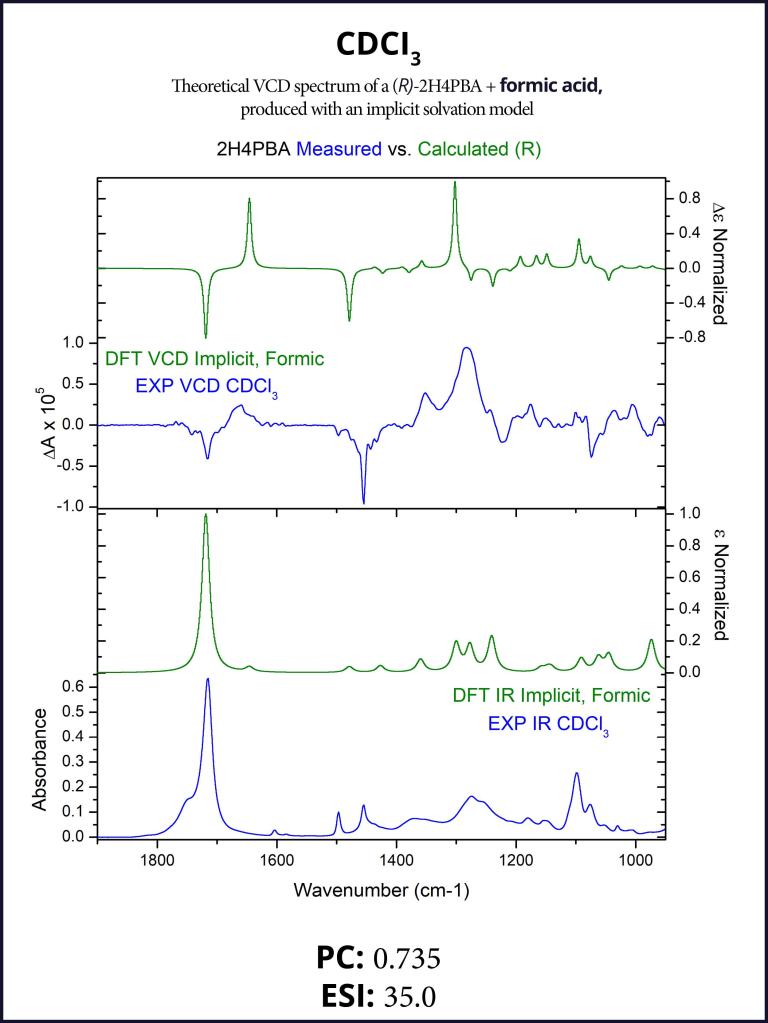
First, we attempt to model the spectra of (R)-2H4PBA in an implicit solvent. Figure 1 below shows the theoretical and experimental infrared (IR) and VCD spectra of (R)-2H4PBA. The left panel corresponds to CDCl3 whereas the right one represents DMSO-d6. Deuterated solvents are used for experimental spectra to avoid overlap of solvent IR absorption bands with those of the sample.
The theoretical VCD calculations using an implicit solvation model applied to (R)-2H4PBA yield a reasonable agreement with the experimental data for both solvents such that absolute configuration can be assigned. The Pearson coefficients (PCs) of the spectral overlap between the experimental and theoretical VCD spectra are shown below the spectra. PCs with the absolute value above 0.2 are normally sufficient to provide confidence to stereochemistry assignment,3 so the coefficients of 0.776 and 0.618 confirm that the visual assignment of the stereocenter as R is correct. The ESI4 also favors the assignment of R for the absolute configuration. Just as in the case for PCs, ESI has a lower value for the solvent DMSO-d6, compared to its value for CDCl3 . These lower values of DMSO-d6 indicate a lower level of overlap between theory and experiment.
One clearly visible qualitative disagreement between theory and experiment is the carboxyl band at around 1700 cm-1 for both solvents. The theoretical frequency is almost always shifted in this region due to lack of accounting for intramolecular or intermolecular hydrogen bonding. In the case of acid, as is here, this is most likely due to the molecule dimerization in CDCl3 involving the COOH group and solvent-solute hydrogen bonding in DMSO.
It is known that DMSO can form hydrogen bonds with some solutes, altering its conformational landscape.5 Such hydrogen bonds must be formed between 2H4PBA, which contains a hydroxyl-group, and DMSO. This is probably the main reason why the experimental VCD spectra in chloroform and DMSO are different. Theoretical spectra of 2H4PBA calculated in implicit chloroform/ DMSO solvent are not able to account for hydrogen bond formation and so, quite predictably, are very similar.
Figure 1: FTIR (lower panels) and VCD (upper panels) comparison between experimental spectra (blue) and spectra simulated in an implicit solvent (green). Some qualitative disagreement, particularly around 1700 cm-1, can be observed.
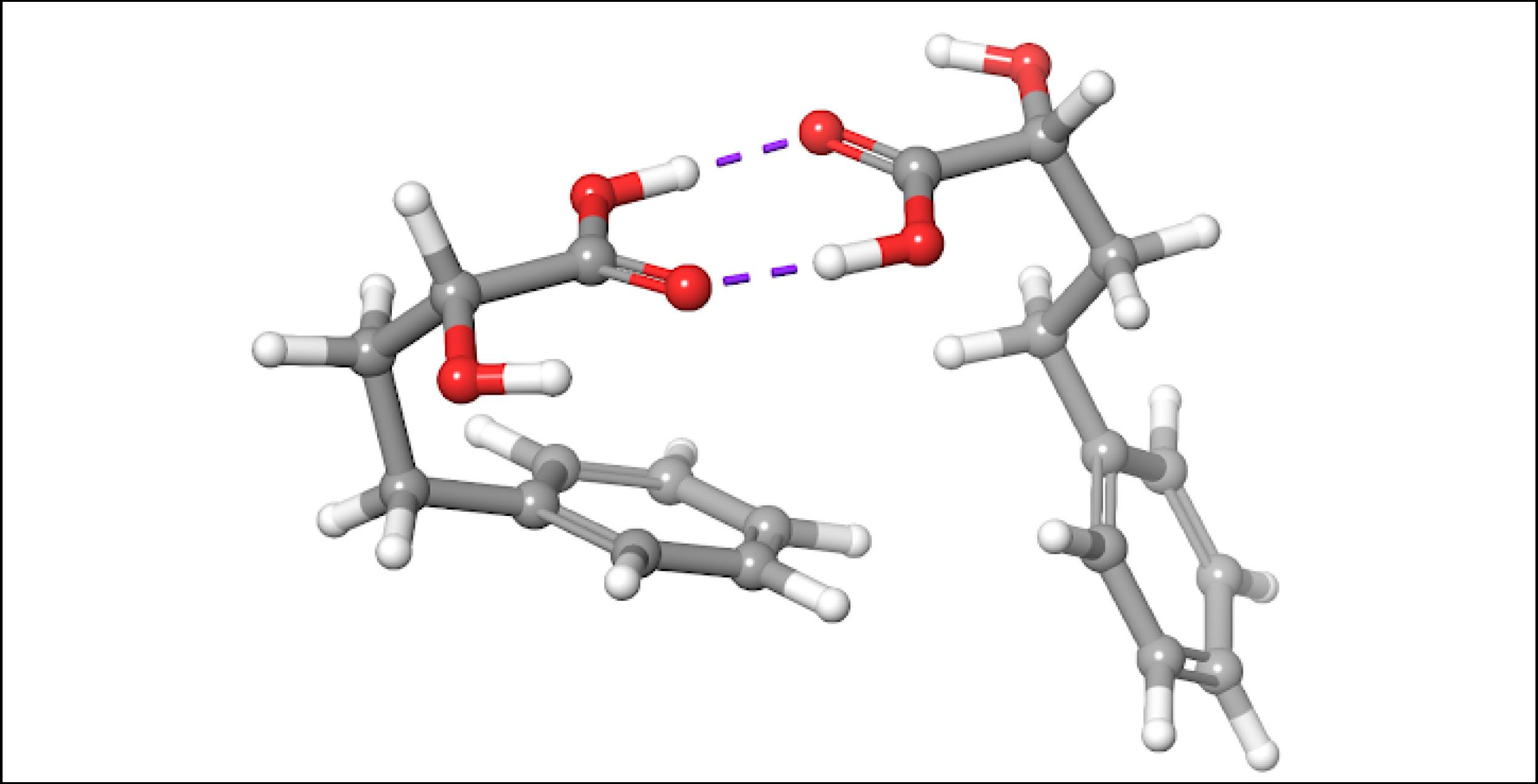
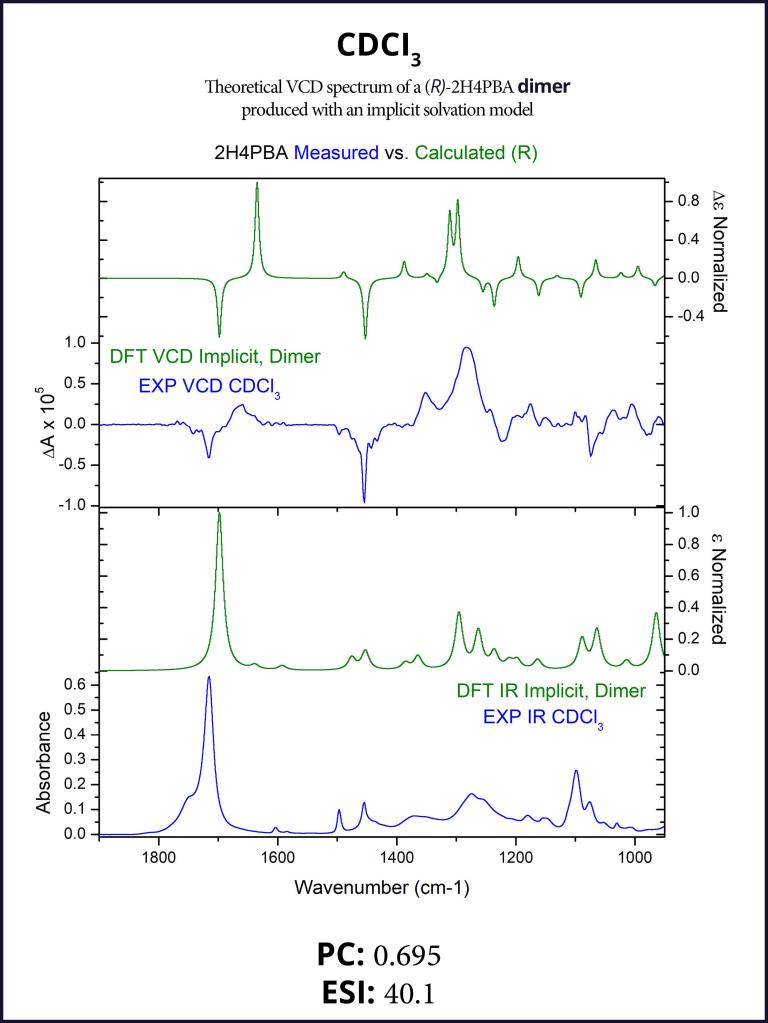
Obtaining a better spectral overlap in chloroform
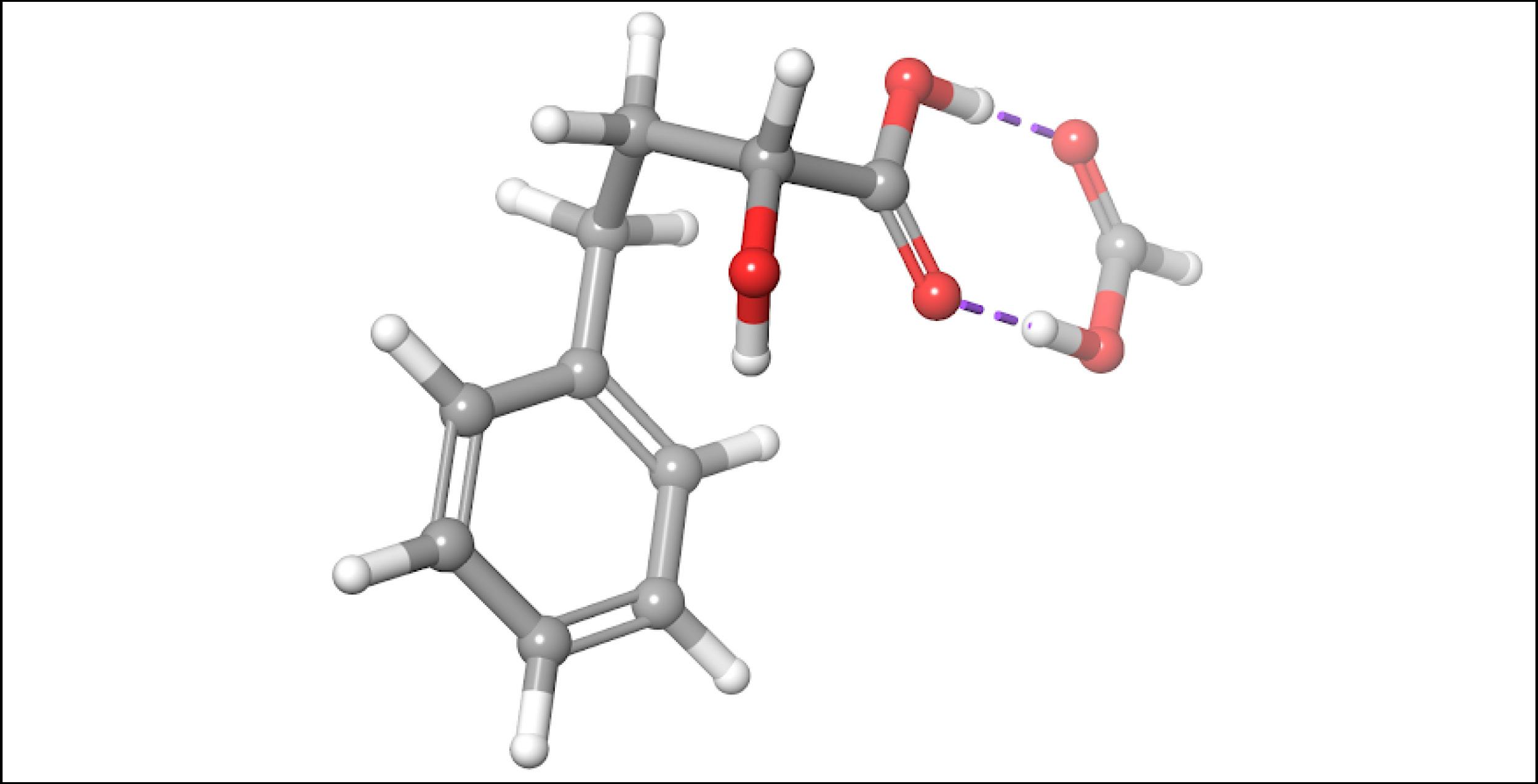
To account for the likely dimerization of (R)-2H4PBA in chloroform, we conducted a similar VCD calculation on a (R)-2H4PBA dimer (see Figure 2) using the same implicit solvent. The resulting predicted spectra show a much better visual agreement with the experimental spectra (Figure 3). VCD calculations on a dimer are significantly more computationally expensive (such a calculation including conformational search took 2 days on 8 CPUs) than those on a monomer (2 hours on 8 CPUs). One way to minimize the time expense is to ‘mimic’ modeling of the dimer as (R)-2H4PBA in a hydrogen-bonded complex with a much smaller formic acid, as shown in Figure 4. Figure 5 shows that the results of such calculation have a good agreement with the experiment. The resulting spectrum is similar in shape to that of the full (R)-2H4PBA dimer, and has the correct sign of the peaks in the region 1700-1850 cm-1. The calculation on (R)-2H4PBA + formic acid took approximately 6 hours on 8 CPUs.
Although both the dimer and the formic acid calculations improved the visual agreement and thus a confidence in assignment of the absolute configuration, it is interesting to note that both the PC and the ESI values decreased thus illustrating a very important point: PC and ESI values should only be used to confirm a visual assignment of AC. Subsequently, they provide a quantitative measure for the assignment of AC and the degree of VCD spectral agreement. PC and ESI values depend on both VCD band positions, spectral widths and intensity cancellations for neighboring VCD bands of opposite signs. It is not always possible to see whether changes in a calculation lead to improvement or reduction in the values of the PC or ESI values.
Figure 2: The lowest energy conformation of the dimer of (R)-2H4PBA in chloroform, according to the implicit solvation model. Modeling such dimers may improve the description of VCD spectra of carboxylic acids in chloroform.Figure 3: FTIR (lower panels) and VCD (upper panels) comparison between experimental spectra in CDCl3 (blue) and theoretically calculated spectra of a dimer (green). Modeling of the dimerization effect has greatly improved the qualitative agreement between the experimental and theoretical spectra (compare with the left panel of Figure 1).Figure 4: The lowest energy conformation of (R)-2H4PBA complexed with the formic acid in chloroform, according to the implicit solvation model. Modeling the dimerization effect with the small formic acid saves computational time.
Obtaining a better spectral overlap in DMSO
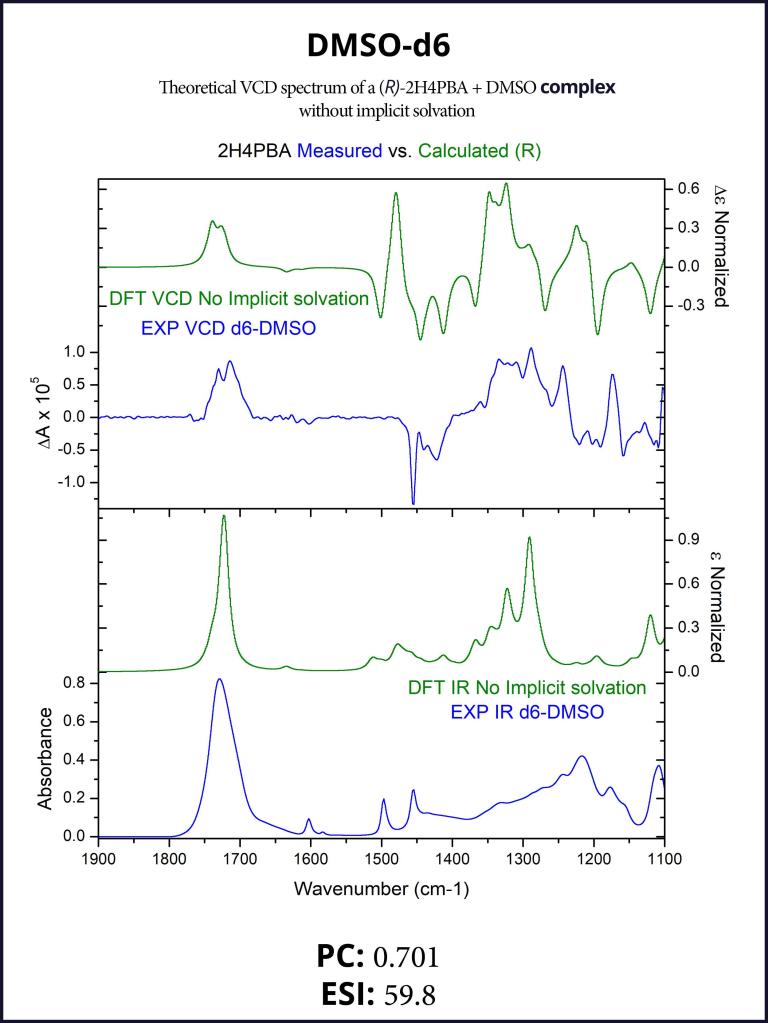
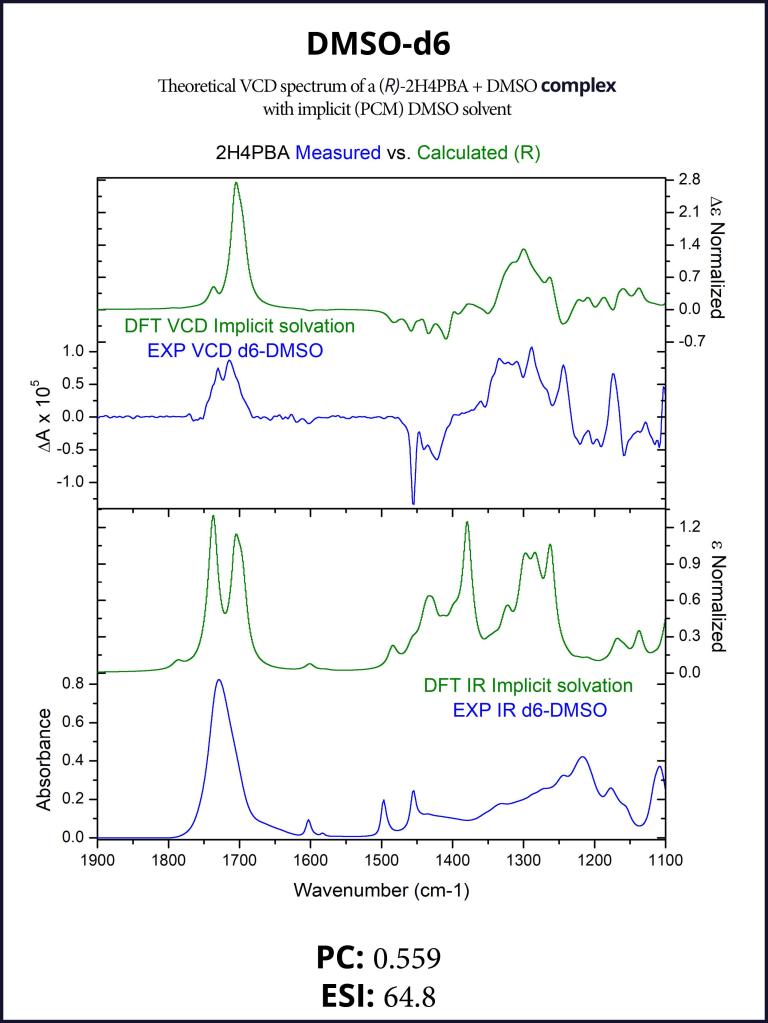
To account for the hydrogen-bonding of (R)-2H4PBA to the solvent DMSO, we set up a complex of (R)-2H4PBA with one molecule of DMSO. We performed VCD calculations on this complex in both the gas phase and with a full solvent PCM shell. The calculation involving an (R)-2H4PBA+DMSO complex with implicit DMSO solvent is consistent with the protocol used for the (R)-2H4PBA dimer in implicit CHCl3. As all the calculations in this work, the VCD calculations included conformational search on the complex.
The implicit DMSO calculations were successful in improving the simulated carboxyl band at around 1700 cm-1. The produced spectra are shown below in Figures 6-7.
Figure 5: FTIR (lower panels) and VCD (upper panels) comparison between experimental spectra in CDCl3 (blue) and theoretically calculated spectra of a complex with formic acid (green). Modeling a smaller complex with formic acid reproduces most spectral features present in the spectra of the full dimer (compare with Figure 3).Figure 6: FTIR (lower panels) and VCD (upper panels) comparison between experimental spectra in DMSO-d6 (blue) and theoretically calculated spectra of the complex (green). The complexation with an explicit DMSO molecule improves the theoretical VCD spectrum around 1700 cm-1 (compare with the right panel of Figure 1).Figure 7: FTIR (lower panels) and VCD (upper panels) comparison between experimental spectra in DMSO-d6 (blue) and theoretically calculated spectra of the complex with implicit solvation (green). The complexation with an explicit DMSO molecule in an implicit solvent further improves the VCD spectrum in the region 1100-1500 cm-1 (compare with Figure 6 and the right panel of Figure 1).
Conclusions
Determination of absolute configuration can be routinely achieved by combining experimental VCD measurements (BioTools, Inc.) with theoretical VCD calculations (Schrödinger, Inc.). As solvent effects on VCD spectra can be significant it is advisable to model these effects with an implicit solvent model. For a better agreement between experimental and theoretical VCD spectra of some compounds it might be useful to go beyond standard implicit solvent calculations by:
(i) Modeling dimers of compounds prone to forming intermolecular hydrogen bonds in non-polar solvents;
(ii) Using a complex with implicit solvent molecules for a better representation of solute-solvent interactions driven by hydrogen bonding.
Experimental VCD Spectra Measurements
Approximately 8-10 mg of 2H4PBA was dissolved in ~125-250 μL and the resulting solution was transferred to a BaF2 IR cell with path length of 100 μm. Spectra were measured on BioTools’ (Jupiter, Florida) ChiralIR2X DualPEM FT-VCD spectrometer, resolution 4 cm-1, PEM maximum frequency 1400 cm-1. The samples were measured for 8 blocks of 1 hour each while purged with dry air to remove water vapor. IR spectra were processed by solvent subtraction and offset to zero at 2000 cm-1. The VCD blocks were averaged, then processed by half difference method [(E1 – E2) / 2].
Theoretical VCD Spectra Calculations
The theoretical VCD spectra were modeled with the standard computational VCD workflow by Schrödinger, Inc. A MacroModel/OPLS4 conformational search retains up to 100 conformations within 5 kcal/mol energy window. All subsequent DFT calculations are carried out with Jaguar at the B3LYP-D3/LACVP** level of theory. When implicit solvent models are requested, the geometries of the conformations are optimized with PCM (polarizable continuum model) and a final single point energy evaluation for determining Boltzmann weights is done with PBF (Poisson-Boltzmann finite elements). If no implicit solvent is requested, the geometries of the conformations are optimized in the gas phase. VCD spectra of the conformations that fit within 5 kcal/mol energy window are computed with or without implicit PCM solvent, as requested. The spectra of individual conformations are Boltzmann-averaged and then normalized to produce the final VCD spectrum. Alignment of theoretical and experimental spectra is achieved according to the algorithm described in Ref. 3.
Progress in understanding atomic level processing at the atomic scale
Share
Speaker
Simon Elliott
Director – Atomic Level Process Simulation
Abstract
In this talk, we will dip into stories about how simulations have advanced our understanding of the growth mechanisms of ALD, and lately of ALE too. We will also sketch out work on nucleation onto substrates, metal deposition, plasma-surface reactions and precursor design. Building on this, simulations are now showing how to control continuous versus self-limiting processes, and deposition versus etch.
We will emphasize how collaboration has been the main driver of this work, always checking and challenging the relevance of the model results for actual experiments. The wider uptake of atomic-scale modelling as a research tool is confirming its usefulness and impact.
As long as computer power continues to snowball, the future for modelling process chemistry is bright. We are looking forward to finding improved precursors through the semi-autonomous exploration of chemical space. Machine-learning techniques are opening up areas that were out-of-bounds to simulations based on physical laws, such as prediction of complex properties and increased accuracy in interatomic potentials for large systems.
Schrödinger デジタル創薬セミナー: Into the Clinic ~計算化学がもたらす創薬プロセスの変貌~
Share
Speakers
Goran Krilov
Senior Director, Schrodinger Therapeutics Group
Abstract
Mucosa-associated Lymphoid Tissue Lymphoma Translocation Protein 1 (MALT1) is a genetically validated target for the treatment of diseases associated with lymphocyte regulation. Unlike first generation inhibitors, centered on large peptidomimetics targeting the protease domain, second generation inhibitors targeting an allosteric region at the interface of the caspase-like and Ig3 domains are much more promising. Still, significant challenges remain in optimizing properties such as permeability, efflux, and solubility, while maintaining on-target potency. Harnessing the full potential of the Schrödinger platform which combines rigorous physics-based modeling with machine learning (ML), predictive ADMET models, and data analytics to search and triage a chemical space, we were able to rapidly identify multiple novel potent series. Subsequent in-silico multi parameter optimization (MPO) campaign quickly identified SGR-1505, a potential best-in-class MALT1 inhibitor with balanced properties and on-target activity, within 10 months of the start of the project and having synthesized only 129 compounds. SGR-1505 demonstrates strong positive effects in patient-derived B-cell tumor models both as a single agent as well as in combination with existing standard of care, and is currently progressing through Phase I clinical trials.
Performed rapid in silico design cycles using a collaborative platform and a large-scale de novo design workflow
Optimized potency and selectivity with relative binding FEP+ and protein FEP+
Resulted in a development candidate currently in preclinical development
Target
Wee1, Ser/Thr kinase
Program Type
Schrödinger proprietary program, small molecule
Indication
Solid tumors
Stage
Phase 1 clinical trial
“This program demonstrates the first prospective application of protein FEP+ to model broad kinome selectivity, which not only enabled the Wee1 team to rapidly identify a few highly selective chemotypes and rescue a program that had hit a major roadblock, but it also added a powerful tool for the broader drug discovery community to tackle selectivity challenges more efficiently.”
Jiashi Wang
Senior Director, Medicinal Chemistry
Schrödinger Therapeutics Group
Design challenge
Inhibition of Wee1, a serine/threonine protein kinase which serves as the gatekeeper of the G2-M cell-cycle checkpoint, forces cells into unscheduled mitosis and culminates in cell death. Given its critical role in DNA repair, Wee1 is an attractive target for oncology drug development. Clinical trial data for the Wee1 inhibitor adavosertib (AZD-1775) validated the potential of targeting Wee1 by revealing strong anti-cancer activity in solid tumors.1-11 However, it was suspected that AZD-1775 had potential liabilities due to inhibition of several other kinases, including the PLK family, as well as time-dependent inhibition of CYP3A4.
The aim of this program, driven by Schrödinger Therapeutics Group, was to develop a best-in-class, highly selective Wee1 inhibitor by leveraging rigorous physics-based modeling approaches to address off-target liabilities with high precision.
Efficient generation of a selective and potent lead series with a rigorous FEP-based workflow
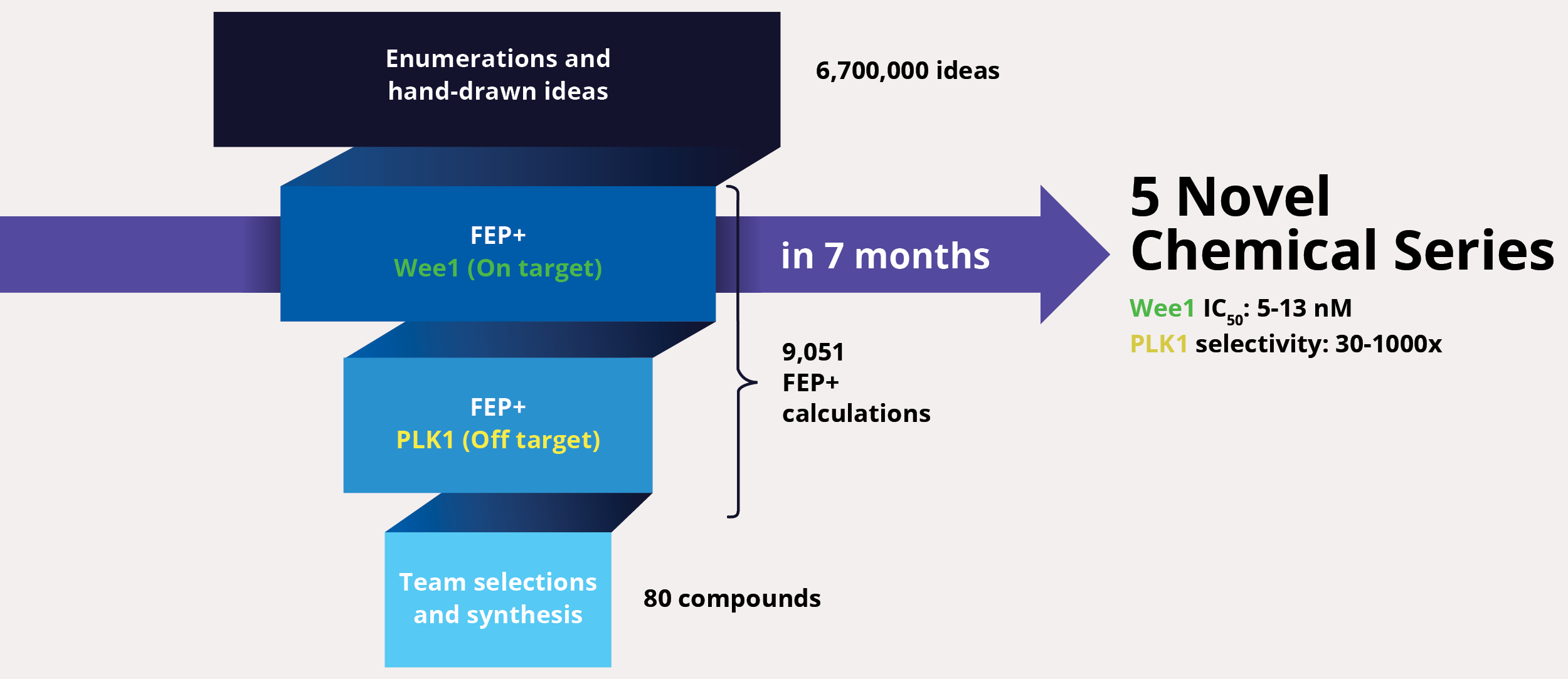
At project onset, the team’s goal was to identify one or multiple novel lead series with improved selectivity for Wee1 as compared to polo-like kinase 1 (PLK1). The team enumerated over six million ideas and hand-drawn designs, then triaged them using an automated chemistry-based filtering workflow, structure-based screening (docking), and ultimately rigorous calculation of relative binding free energies via free energy perturbations (FEP+, De Novo Design Workflow) (Figure 1). The accuracy and utility of FEP+ as a computational binding affinity assay has been validated extensively, generating predictions within 1.0 kcal/mol of experimental values on average.12 By combining FEP+ with high performance cloud computing and machine learning (Active Learning FEP+), over 9,000 FEP+ calculations were performed to evaluate the affinity of the most promising designs for inhibiting Wee1 and ensuring selectivity over PLK1.
Design ideas were then evaluated by the project team in LiveDesign, Schrödinger’s cloud-based enterprise informatics platform. These workflows informed the selection of less than 100 compounds for synthesis and testing, resulting in the identification of multiple novel chemotypes with nanomolar affinity and enhanced selectivity (up to 1000x) for Wee1 over PLK1 within seven months from project start.13
Figure 1: FEP+ workflow to identify potent and selective Wee1 inhibitors, and enhance selectivity for Wee1 over PLK1.
Addressing additional off-target liabilities with protein FEP+
Although the team successfully improved selectivity against PLK1 in the first phase of the project, subsequent kinase panel screening revealed a significant number of unanticipated off-target kinase liabilities. In lieu of starting over or scrapping the program entirely, the team explored a complementary strategy to improve selectivity of the chemotypes identified in the first phase. A major driver of selectivity seemed to be a specific residue in the binding site. The team utilized protein FEP+ calculations, a protocol within FEP+, to assess the impact of single point-mutations at that location on the ligand affinity and to infer selectivity across a large diversity of kinases without the need to profile each kinase separately. In this new workflow, 6,700 new designs were profiled with ligand FEP+ for predicting potency against Wee1 and with protein FEP+ for predicting broad kinome selectivity. During this three month modeling campaign, the ligand FEP+ and protein FEP+ strategy identified 42 promising molecules for synthesis and 22 of these molecules exhibited low nanomolar to picomolar measured potencies against Wee1 with substantially reduced selectivity liabilities (Figure 2).14
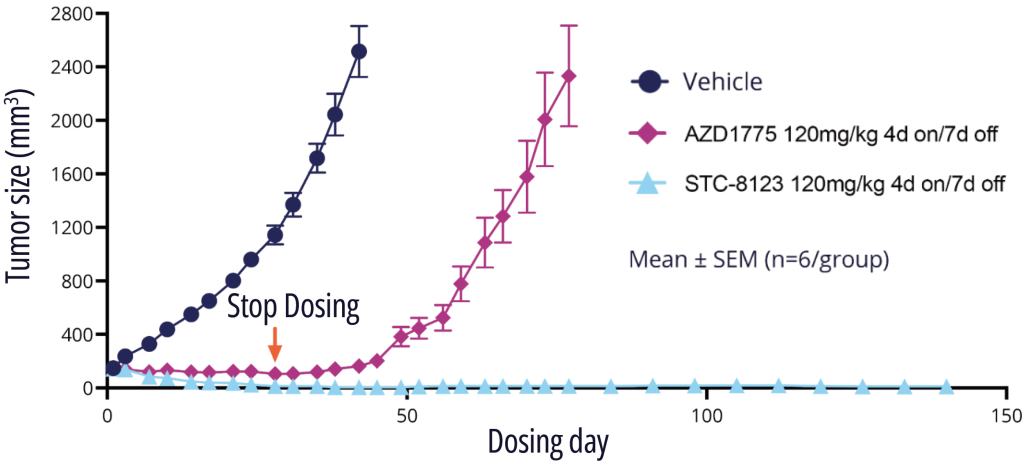
One exquisitely selective molecule, STC-8123, was used as a proof-of-concept compound to demonstrate that highly selective Wee1 inhibitors retain profound in vivo efficacy. Indeed, in an A427 mouse model with intermittent dosing, STC-8123 demonstrated rapid and more sustainable inhibition of tumor growth compared with AZD-1775 (Figure 3).14
Figure 2: Kinome selectivity profiling using scanMAX Kinase Assay Panel confirms that the FEP+/Protein FEP+ workflow enabled the rapid optimization of non-selective hits into multiple series with gene-family wide selectivity — as exemplified by STC-8123.
Figure 3: No tumor regrowth observed after STC-8123 high dose treatment stopped in A427 (non-small cell lung adenocarcinoma) tumor model.14
Optimizing DMPK and ADME properties with machine learning and quantum mechanics strategies
With a promising chemical series in hand, the team began the process of optimizing DMPK and ADME properties. As experimental data accumulated for the lead series, the team trained project-specific machine learning (ML) models to predict time-dependent inhibition of CYP3A4 profiles as well as a variety of ADME properties (DeepAutoQSAR). In addition, quantum mechanical (QM) calculations were developed for modeling compound reactivity with the CYP3A4 heme, a potential target for drug-drug interaction liabilities (Jaguar). During late-stage lead optimization, this breadth of ML models and QM physics-based calculations enabled prospective multiparameter optimization to readily narrow down the chemical space that was sent for synthesis and testing.
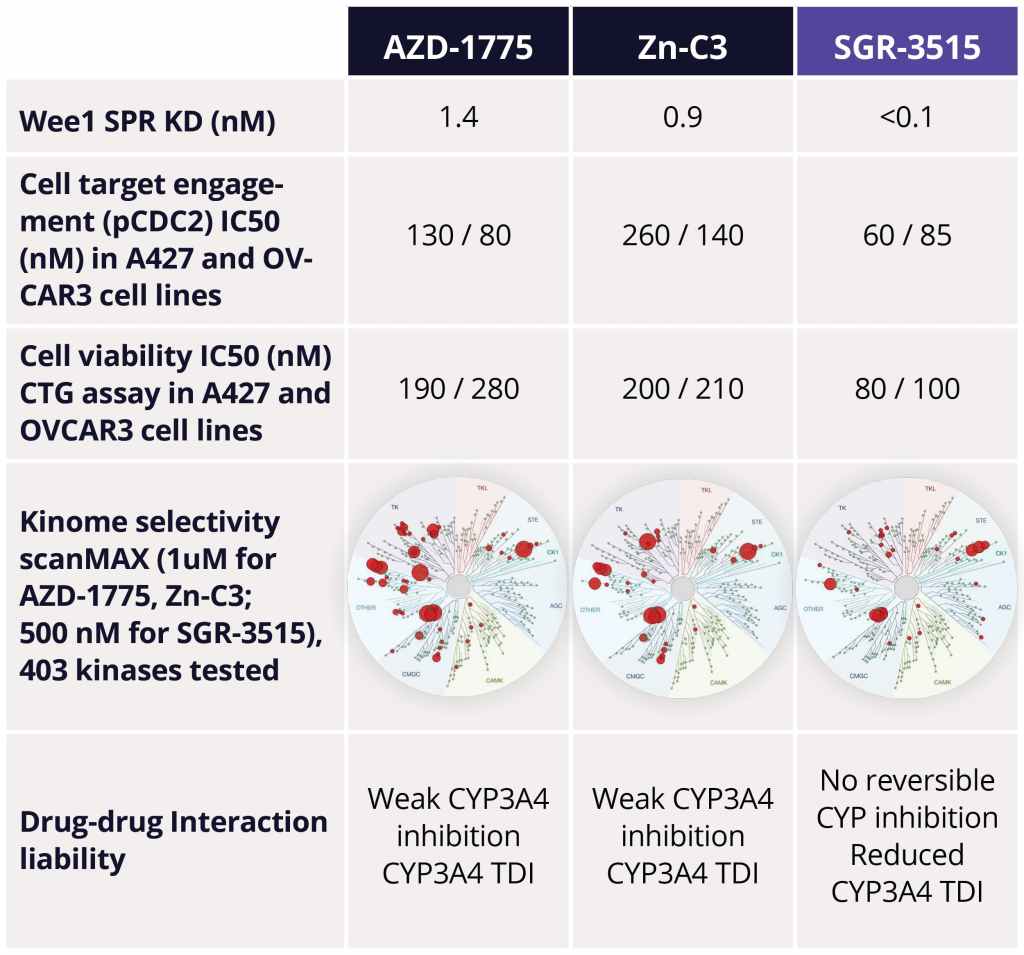
The team identified several advanced leads from among the ~300 compounds synthesized in the series. Upon in-depth profiling of advanced leads and careful consideration of the properties that a best-in-class, next generation Wee1 inhibitor should have, SGR-3515 was nominated as the development candidate. SGR-3515 is an exquisitely selective, potent molecule that is structurally-differentiated from competitors’ molecules, has a differentiated ADME profile, and achieves superior in vivo efficacy (Figure 4).
Figure 4: Comparison of SGR-3515 with competitors’ Wee1 inhibitors.15 SGR-3515 shows superior binding affinity and potency in cellular assays in A427 and OVCAR3 (high-grade serous ovarian adenocarcinoma) cell lines, demonstrates excellent broad kinome selectivity, and reduces drug-drug interaction potential due to decreased time-dependent inhibition (TDI) of CYP3A4. All competitor data was internally generated by contract research organizations.
Enabling digital technologies to drive discovery programs
FEP+
Elucidation of ligand binding preferences for large families of off-targets with protein FEP+ and on-target potency with FEP+
Cancer Res (2019) 79 (13_Supplement): CT02. 12.J Clin Oncol 2021 Nov 20;39(33).
Advancing drug discovery through enhanced free energy calculations. Abel et al.
Acc. Chem. Res. 2017, 50, 7, 1625–1632.
De-risking off-target liabilities with protein free energy methods.
Knight et al. ACS 2022.
Discovery of potent, selective, and orally available WEE1 inhibitors that demonstrate increased DNA damage and mitosis in tumor cells leading to tumor regression in vivo.
Sun et al. AACR 2022.
Into the clinic: Transforming the drug discovery process with digital chemistry.
Davis et al. Lab of the Future 2023.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.