Design, develop, and optimize the next generation of automotive materials at the molecular level
Leverage molecular simulation and machine learning to guide in silico design of novel materials that meet sustainability requirements and increase efficiency.
Brilliant lighting and displays
Develop efficient, durable, and low-cost organic electronic materials for lighting and display systems.
Accelerate innovation with sustainable ingredients. Computationally screen formulations for paints, coatings, and lubricants while optimizing for improved durability and efficiency.
Rapidly uncover lightweight, durable, and high-performing polymers, while optimizing manufacturing by digitally investigating the root causes of failure and defects.
Discover how Schrödinger technology is being used to solve real-world research challenges.
Materials Science Webinar
Accelerating product development with computational materials engineering
Learn how Ansys and Schrödinger are transforming product development with Integrated Computational Materials Engineering (ICME) to accelerate material discovery and innovation.
Advancing battery materials innovation using charge-aware machine learning force fields
In this webinar, we will demonstrate how Schrödinger is utilizing an integrated computational approach combining physics-based molecular modeling with machine learning force fields (MLFFs) to address key challenges in battery materials design.
Accelerating materials discovery with physics-informed AI/ML
This webinar series will explore how cutting-edge computational methods are revolutionizing the design and optimization of pharmaceutical drugs, biologics , and advanced materials.
Advancing machine learning force fields for materials science applications
In this webinar, we will introduce Schrödinger’s state-of-the-art MLFF architecture, called Message Passing Network with Iterative Charge Equilibration (MPNICE), which incorporates explicit electrostatics for accurate charge representations.
Accelerating chemical innovation with AI/ML: Breakthroughs across materials applications
In this webinar, we will explore how AI/ML is driving impactful advancements in materials innovation, highlighting case studies that illustrate cutting-edge ML techniques in diverse applications.
High-performance materials discovery: A decade of cloud-enabled breakthroughs
This talk will showcase how Schrödinger’s integrated materials science platform enables massive parallel screening and de novo design campaigns across diverse applications.
How Physics-based Modeling and Machine Learning Enable Accelerated Development of Battery Materials
In this webinar, we focus on examples to demonstrate the application of automated solutions for accurate prediction of thermodynamic stability and voltage profile of cathode materials, ion diffusion pathways and kinetics in electrode materials, transport properties of liquid electrolytes and modeling the nucleation and growth of solid electrolyte interphase (SEI) layers using Schrödinger’s SEI simulator module.
AI/ML meets physics-based simulations: A new era in complex materials design
In this webinar, we demonstrate the application of this combined approach in designing materials and formulations across diverse materials science applications, from battery electrolytes and fuel mixtures to thermoplastics and OLED devices.
Schedule a consultation on Schrödinger’s automotive and transportation solutions.
Contact us today to explore how you can leverage advanced simulation and AI/ML for automotive and transportation.
Don’t see your areas of interest above? Reach out so we can help.
Form submitted
Thank you, we’ll be in touch soon.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Maestro is Schrödinger’s streamlined portal for access to state-of-the-art predictive computational modeling and machine learning workflows for molecular discovery. With an intuitive, advanced graphical user interface, Maestro provides users of all experience levels a unified entry point for gaining novel molecular insights to drive their research.
Advantages of Maestro for molecular design and discovery
Easy-to-use graphical interface
Create models and analyze results with a simple, guided graphical user interface and step-by-step workflows
Decades of innovation at your fingertips
Access technology backed by 30+ years of scientific R&D and validated by thousands of customers across the globe
Fully integrated portal
Benefit from a unified entry point to a wide range of molecular simulation technologies, accessible for users of all experience levels
Power your drug discovery with an integrated platform
Access broad molecular modeling and machine learning capabilities
Connect to a diversity of industry-leading computational tools through a single intuitive interface
Benefit from workflows that are easily searchable and anticipate next steps in common workflows
Model and interpret molecular interactions that aid in design
Reveal structural insights interactively through linked workspace and analysis panels by simply selecting atoms
Read molecules in multiple formats and generate design ideas to facilitate molecular exploration
Easily export models for team-wide collaborative molecular design
Eliminate data silos and improve team collaboration with tightly coupled digital solutions
Import/export structures and models between Maestro and LiveDesign to streamline the discovery process
Simplify your infrastructure by accessing Maestro on the cloud
Make Maestro accessible to teams in a secure, virtual cloud environment
Easily scale compute resources to meet your demands
Creating a Coarse-Grained Model for Protein Formulations
Learn to use the Coarse-Grained Force Field Builder to automatically fit parameters to the Martini coarse-grained force field for a complex protein solution system.
Discover how Schrödinger technology is being used to solve real-world research challenges.
Life Science Webinar
Building a biotech: Enabling a successful digital drug discovery program with a connected platform
Join us to see how this powerful solution can accelerate your DMTA cycles and enable your teams – this isn’t about complex simulations, it’s about giving your team the tools they need to make better decisions, faster.
Building a biotech: Enabling a successful digital drug discovery program with a connected platform
Join us to see how this powerful solution can accelerate your DMTA cycles and enable your teams – this isn’t about complex simulations, it’s about giving your team the tools they need to make better decisions, faster.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Discover better quality molecules, faster with FEP+
FEP+ is Schrödinger’s proprietary, physics-based free energy perturbation technology for computationally predicting protein-ligand binding at an accuracy matching experimental methods, across broad chemical space.
Explore vast chemical space and reduce costs
Leverage FEP+ as an accurate in silico binding affinity assay to drive rapid virtual design cycles and focus experimental efforts on only the highest quality ideas
Improve molecular profiles, efficiently
Optimize multiple properties simultaneously, including potency, selectivity, and solubility, to improve the profile and developability of small and large molecules
Pursue novel chemistry with confidence
Synthesize novel and challenging chemistry with a high degree of confidence through prospective application of FEP+
Continuously pushing the state of the art in free energy methods
Gold standard accuracy
Predictive accuracy approaching experiment (1 kcal/mol) as demonstrated in large-scale validation studies across diverse ligands and protein classes
Proven impact in drug discovery
Widely adopted by leading pharma and biotech companies, with several drug candidates in the clinic driven by FEP+
Highly versatile
Supports the broadest range of applications and perturbation types common in drug discovery scenarios and consistently expanded through active R&D
Apply FEP+ to diverse applications across the drug discovery process
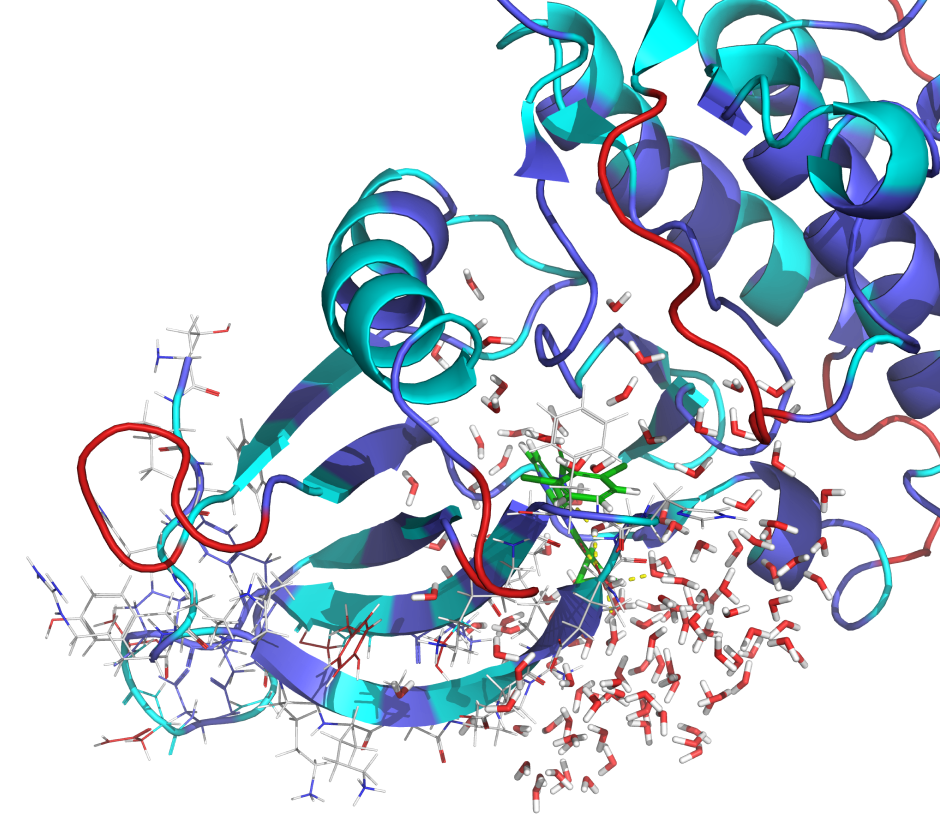
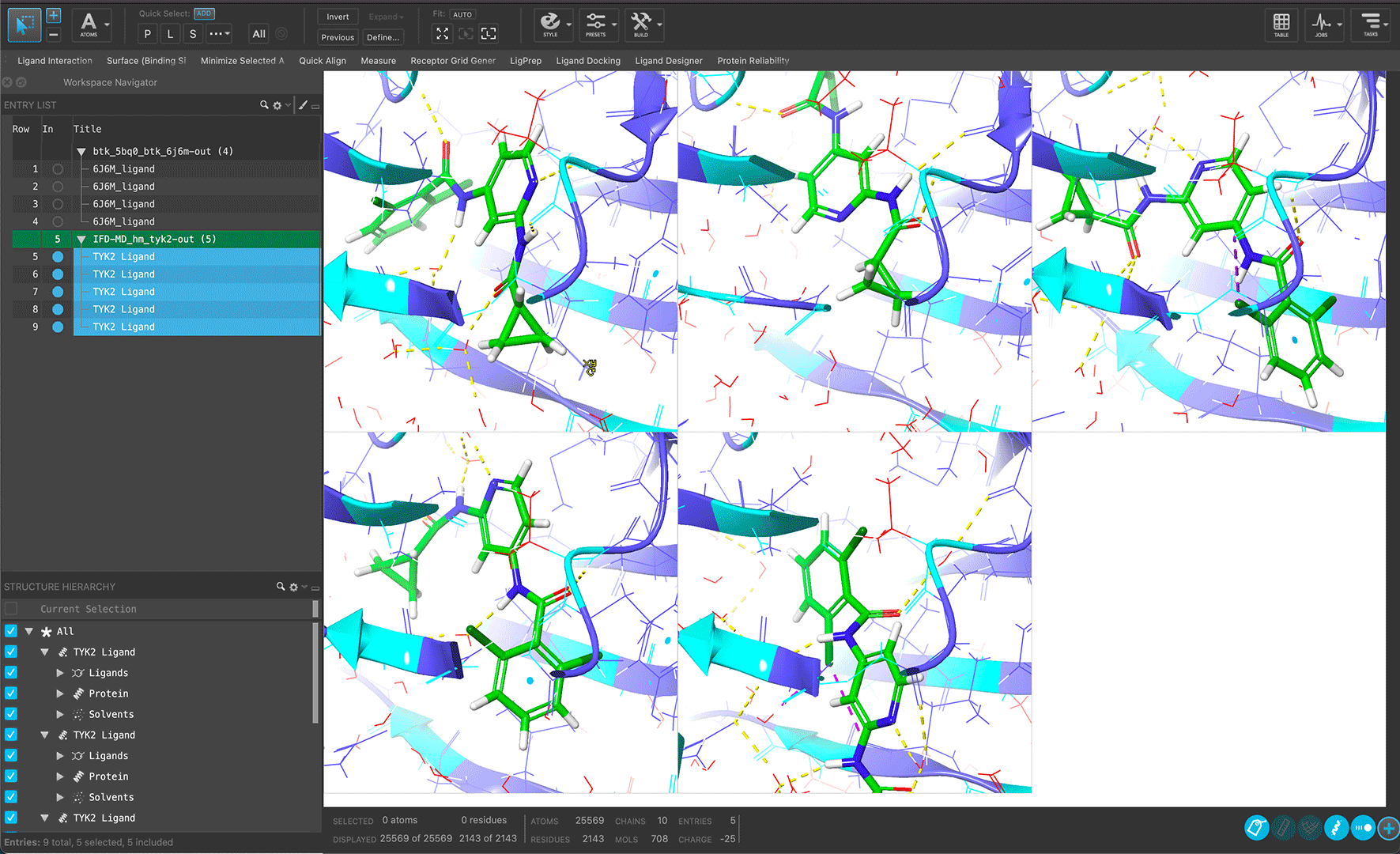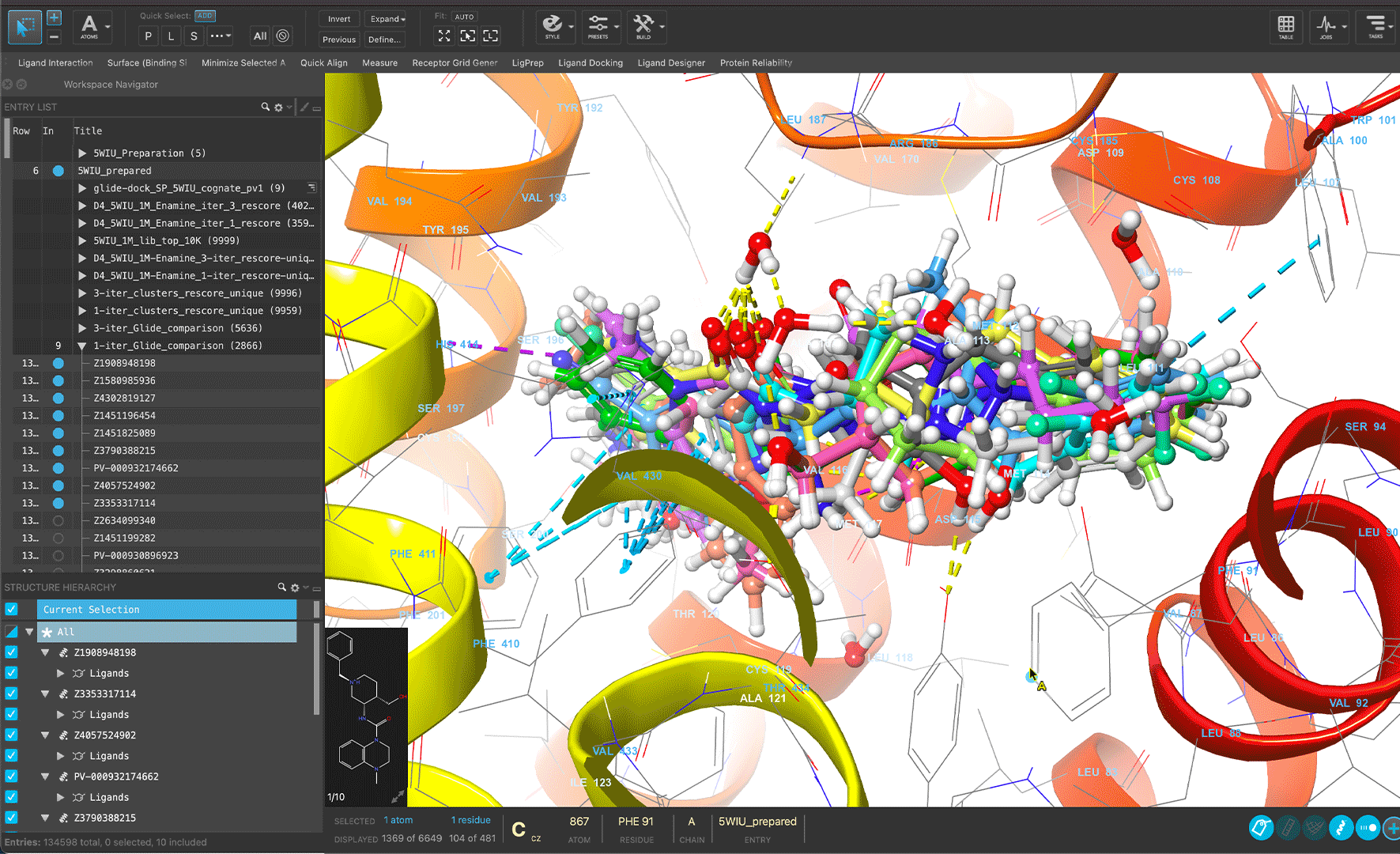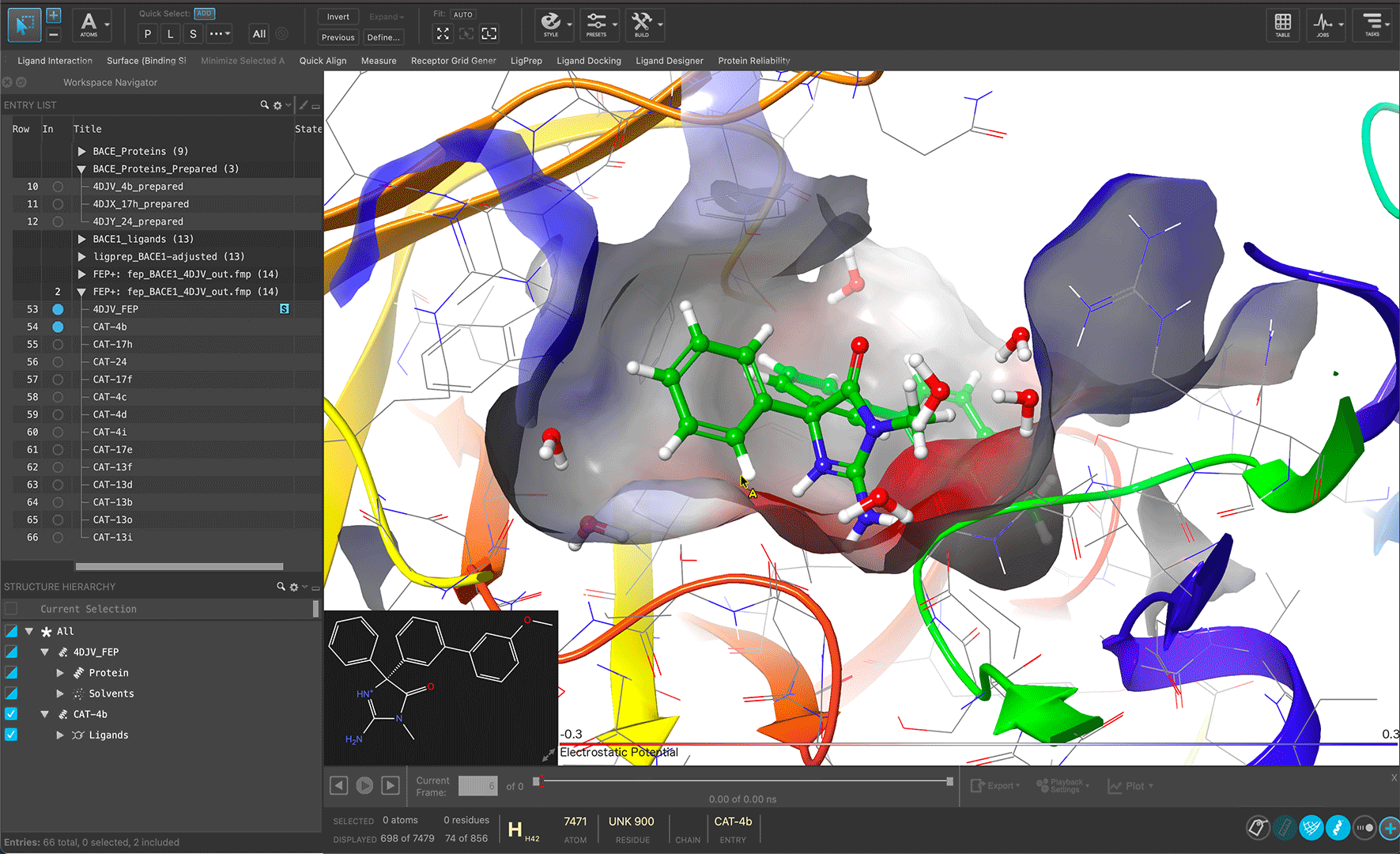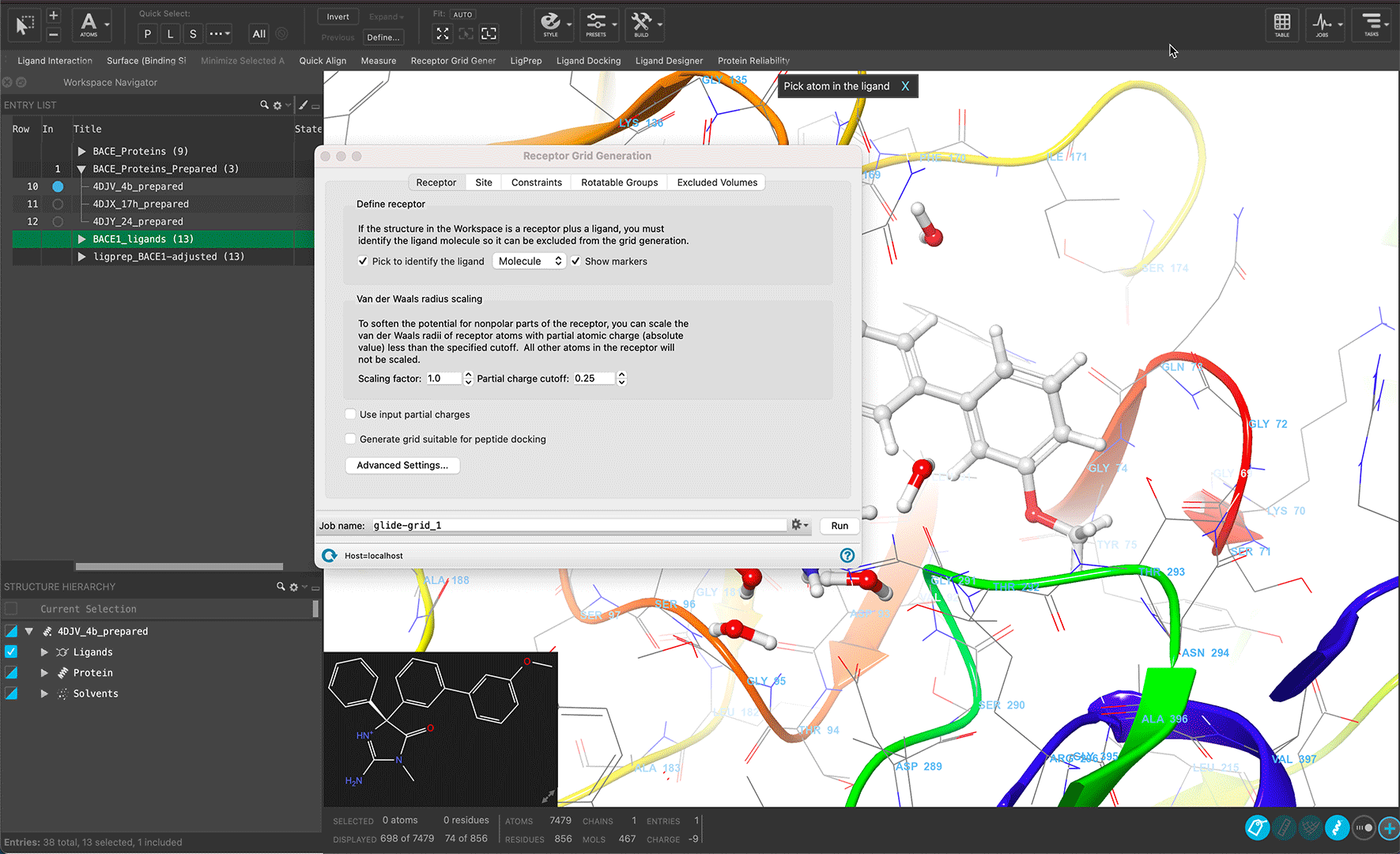
Validate protein models without experimental structures or from low resolution structures using IFD-MD with FEP+
Structurally enable off-targets and design out common ADMET liabilities
Hit Discovery
Rescore hits from virtual screens to prioritize synthesis lists and improve using absolute binding FEP+
Leverage available chemical matter to efficiently discover novel cores via core hopping
Perform large-scale in silico fragment screens using absolute binding FEP+ and solubility FEP+
Hit-to-Lead & Lead Optimization
Rapidly optimize on-target potency by leveraging FEP+ as an in silico binding affinity assay
Optimize selectivity to known off-targets and across large gene families
Maintain on-target potency and selectivity while optimizing ADMET properties
In Silico Protein Engineering
Refine antibody candidate selection with accuracy that reproduces experimentally determined relative free energies
Predict binding affinity, selectivity, and thermostability of peptides
Engineer enzymes for substrate selectivity and specificity
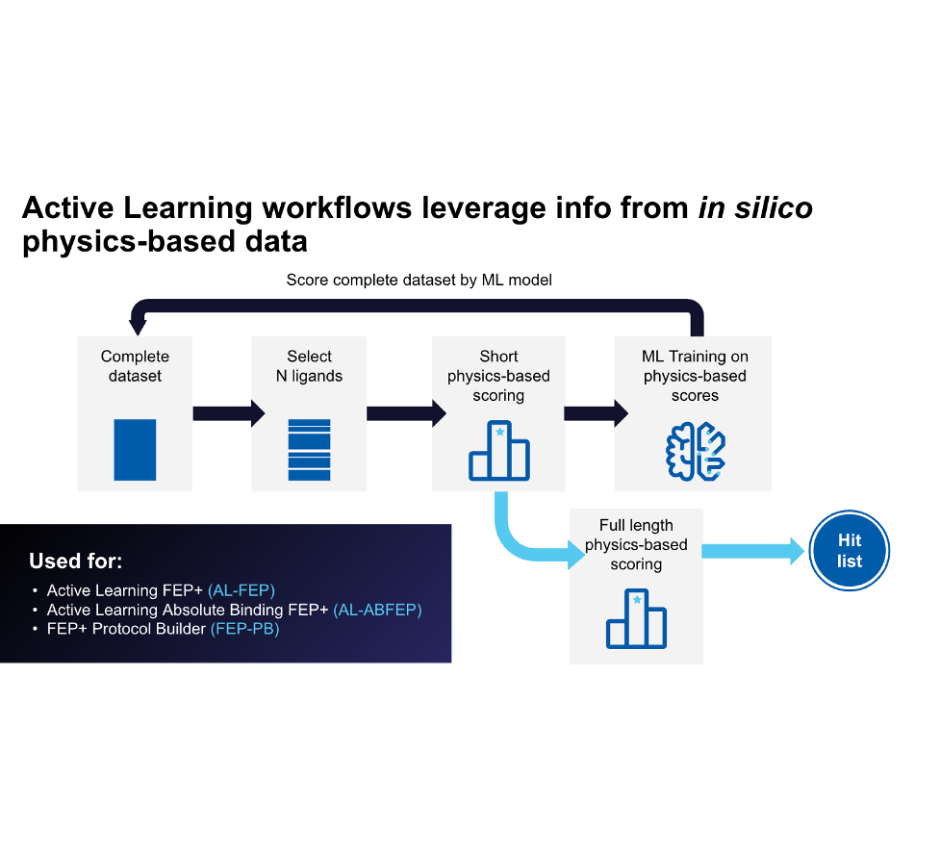
Accelerate FEP+ calculations across large compound libraries with Active Learning
Leverage a well-validated, automated workflow which trains a machine learning model on project-specific FEP+ data to allow processing of up to millions of compounds with highly accurate FEP+ calculations efficiently.
MAR 18, 2026 | Rethinking the rules: Exploiting solvent exposed salt-bridge interactions with free energy perturbation simulations for the discovery of potent inhibitors of SOS1
Diverse computational strategies enable the discovery of p38α-MK2 molecular glues
In this webinar, Schrödinger’s medicinal and computational chemists will show how they used a multipronged computational design strategy to discover multiple structurally diverse, potent, and highly selective molecular glues.
FEP+ Pose Builder — maximizing utility and productivity in FEP simulations
FEP+ Pose Builder is a methodological advancement introduced as an integrated feature to drastically enhance accessibility, user-friendliness, and productivity within the FEP+ pipeline.
Scaling FEP+ for success: Strategic deployment of FEP+ and AI/ML to accelerate chemical space exploration
Join us to map out your strategy for maximizing the organizational impact of FEP+ and to achieve the full potential of your computational drug discovery and business goals.
Rethinking the rules: Exploiting solvent exposed salt-bridge interactions with free energy perturbation simulations for the discovery of potent inhibitors of SOS1
In this webinar, we will walk you through the SOS1 program, as well as our exploration of other examples where these salt-bridge interactions are influential.
FEP+ State of the Union: Advancing computational rigor and scaling predictivity in drug discovery
In this webinar, Robert Abel, Schrödinger’s chief scientific officer, and Schrödinger’s FEP+ experts will provide an in-depth analysis of FEP+’s latest accuracy benchmarks and its expanding domain of applicability, maintaining its position as the gold standard in the industry.
Computational prediction of protein-ligand binding using physics-based free energy perturbation technology at an accuracy matching experimental methods.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Expand your compound design strategies with unbiased chemical space exploration for hit-to-lead and lead opt
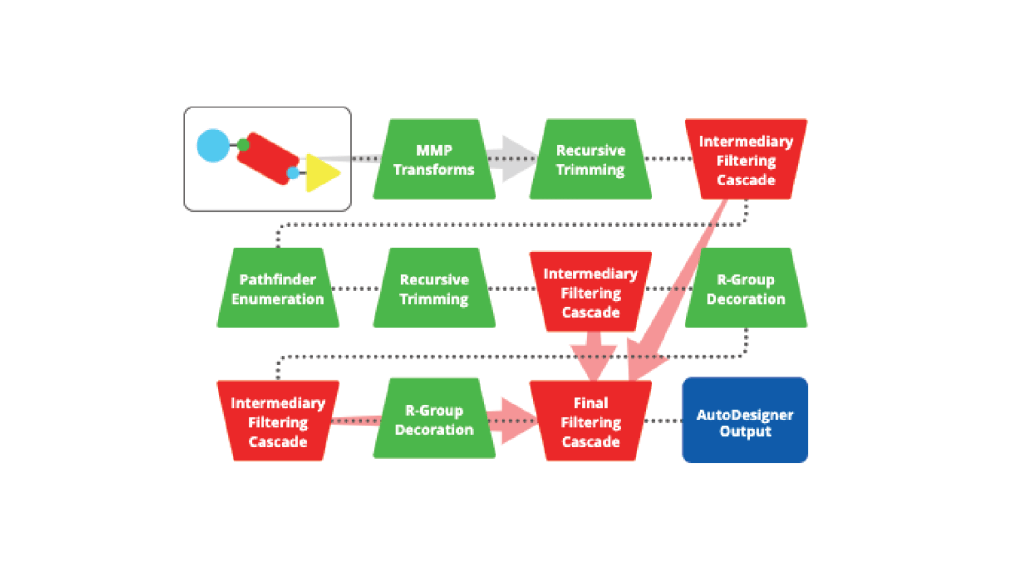
Schrödinger’s De Novo Design Workflow is a fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement. Starting from a hit molecule or lead series, the technology identifies synthetically tractable molecules that meet key project criteria by combining multiple compound enumeration strategies with an advanced filtering cascade (AutoDesigner) and rigorous potency scoring with free energy calculations (Active Learning FEP+).
Key Capabilities
Dramatically improve synthetic tractability of the identified molecules
Through built-in reaction-based enumeration combined with advanced filtering to rule out undesired and unrealistic chemistry
Efficiently identify potent lead compounds in favorable physicochemical property space
By leveraging accurate potency predictions combined with active learning
Fast-track ligand optimization and program success
By efficiently evaluating up to billions of project-relevant virtual molecules
Accelerated, seamless exploration of large chemical space
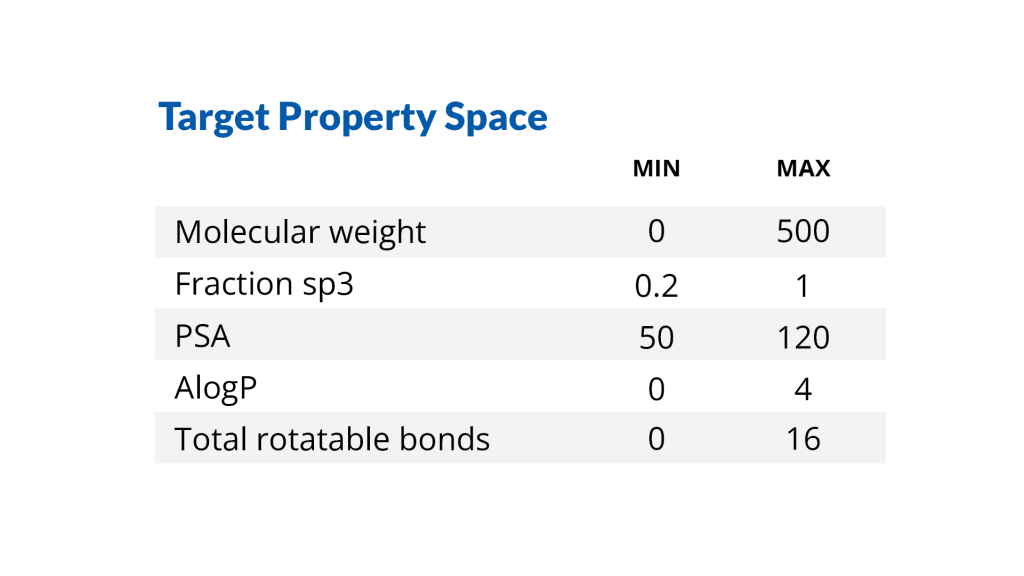
The De Novo Design Workflow offers a cloud-deployable solution with the flexibility to customize settings and property space for the unique needs of your program.
1. Control the chemical space to be explored with project-specific input parameters
Define the starting molecule, the portion of the molecule to explore, the desired physicochemical property space, and additional project-specific filters within LiveDesign, a web-based enterprise molecular design and collaboration platform.
2. Expand into synthetically-tractible space of interest to medicinal chemists
Automatically carry out successive rounds of compound generation and filtering within desired chemical space using cloud-native, multi-stage enumeration strategies combined with an advanced filtering cascade based on physical properties, amenability to FEP+, IP, and docking.
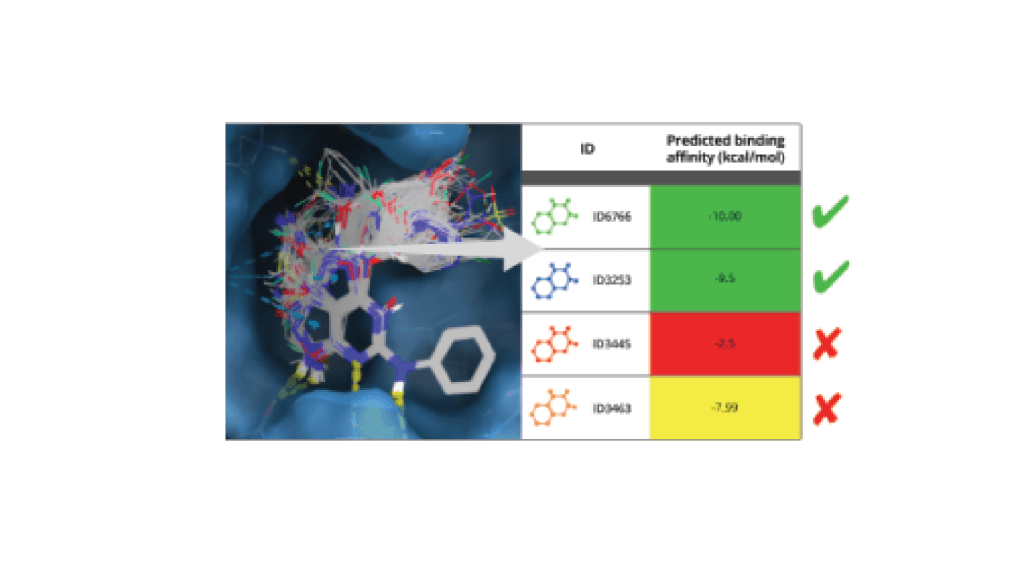
3. Score idea molecules with a highly accurate in silico binding affinity assay
Leverage a well-validated, automated workflow which trains a machine learning model on project-specific FEP+ data to allow processing of up to millions of compounds with highly accurate FEP+ calculations efficiently.
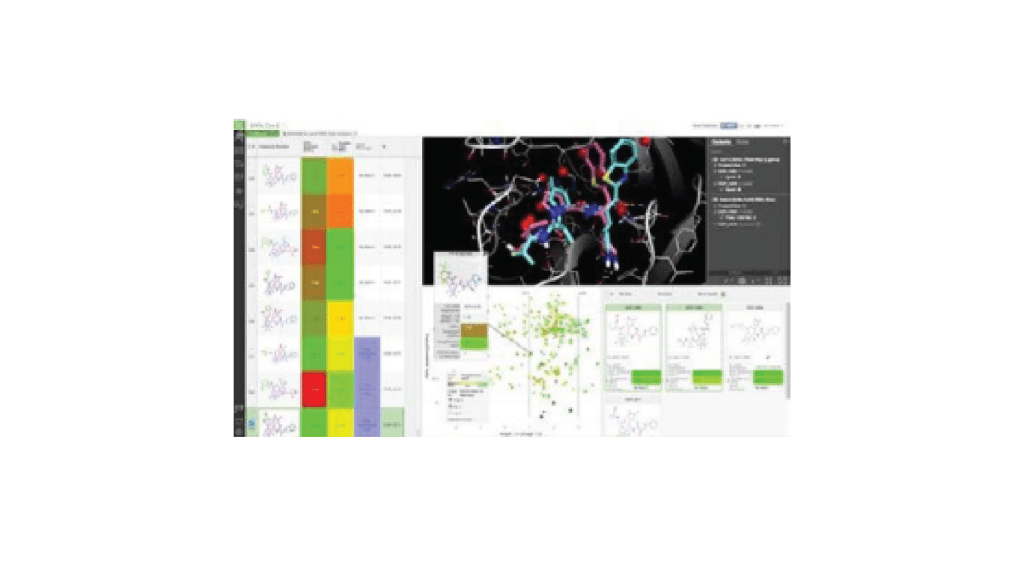
4. Analyze and prioritize output molecules with a collaborative design platform
Review the top scoring compounds and use the FEP+-trained machine learning model in LiveDesign — allowing evaluation, interactive optimization, and prioritization by the project team.
Publications
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Life Science Publication
Accelerated in silico discovery of SGR-1505: A potent MALT1 allosteric inhibitor for the treatment of mature B-cell malignancies
AutoDesigner – Core Design, a De Novo Design Algorithm for Chemical Scaffolds: Application to the Design and Synthesis of Novel Selective Wee1 Inhibitors
AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of D-Amino Acid Oxidase Inhibitors
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Molecular and periodic quantum mechanics, all- atom molecular dynamics, and coarse-grained approaches for studying active pharmaceutical ingredients and their formulations
When registering for the course, you will be able to choose your preferred start and end date. Within those dates, you will have asynchronous access to the course to work on your preferred schedule
Overview
Computational molecular modeling tools have proven effective in materials science research and development. Chemists, physicists and engineers working in materials science will increasingly encounter molecular modeling throughout their careers, making it critical to have a foundational understanding of the cutting edge tools and methods. These courses are ideal for those who wish to develop professionally and expand their CV by earning certification and a badge.
These computational chemistry courses offer an effective and efficient approach to learn practical computational chemistry for materials science:
Work hands-on with Schrödinger’s industry-leading Materials Science Maestro software
Jump start your research program by learning methods that can be directly applied to ongoing projects
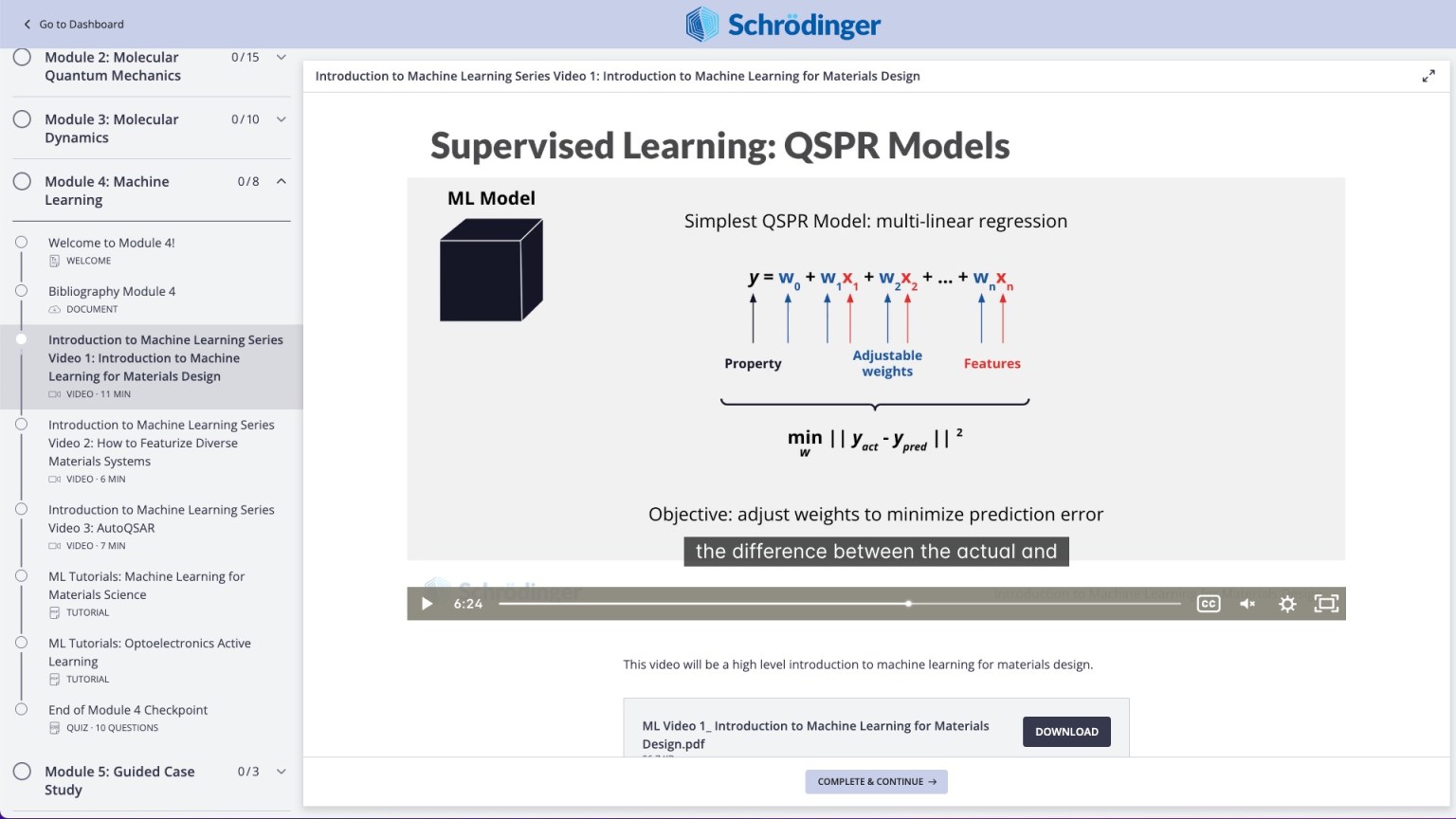
Learn topics ranging from density functional theory (DFT) to molecular dynamics to machine learning for materials design
Perform a completely independent case study to demonstrate mastery of the course content
Benefit from review and feedback from Schrödinger Education Team experts for course assignments and course-related queries
Work on the course materials on your own schedule whenever convenient for you
This course comes with access to a web-based version of Schrödinger software with the necessary licenses and compute resources for the course:
Requirements
A computer with reliable high speed internet access (8 Mbps or better)
A mouse and/or external monitor (recommended but not required)
Working knowledge of general chemistry
Certification
A certificate signed by the Schrödinger course lead
A badge that can be posted to social media, such as LinkedIn
What you will learn
MS Maestro interface
Learn how to use an industry-leading interface for materials science modeling. No coding or scripting required to run modeling workflows
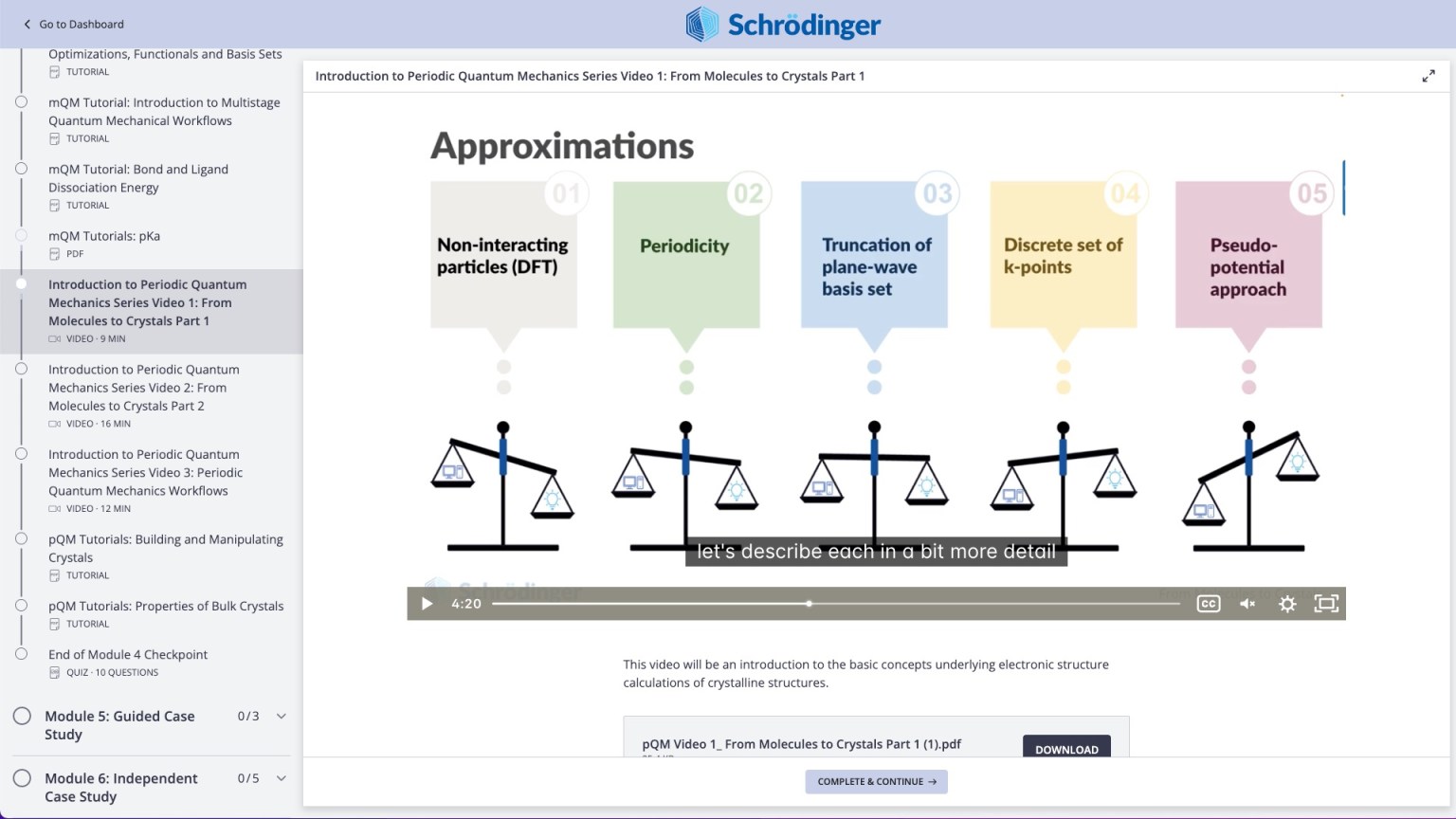
Molecular and periodic quantum mechanics
Learn to apply molecular and periodic density functional theory (DFT) for automated property prediction for amorphous and crystalline active pharmaceutical ingredients
Molecular dynamics
Learn to leverage all-atom MD simulations for simulating properties of complete formulations including miscibility and hygroscopicity
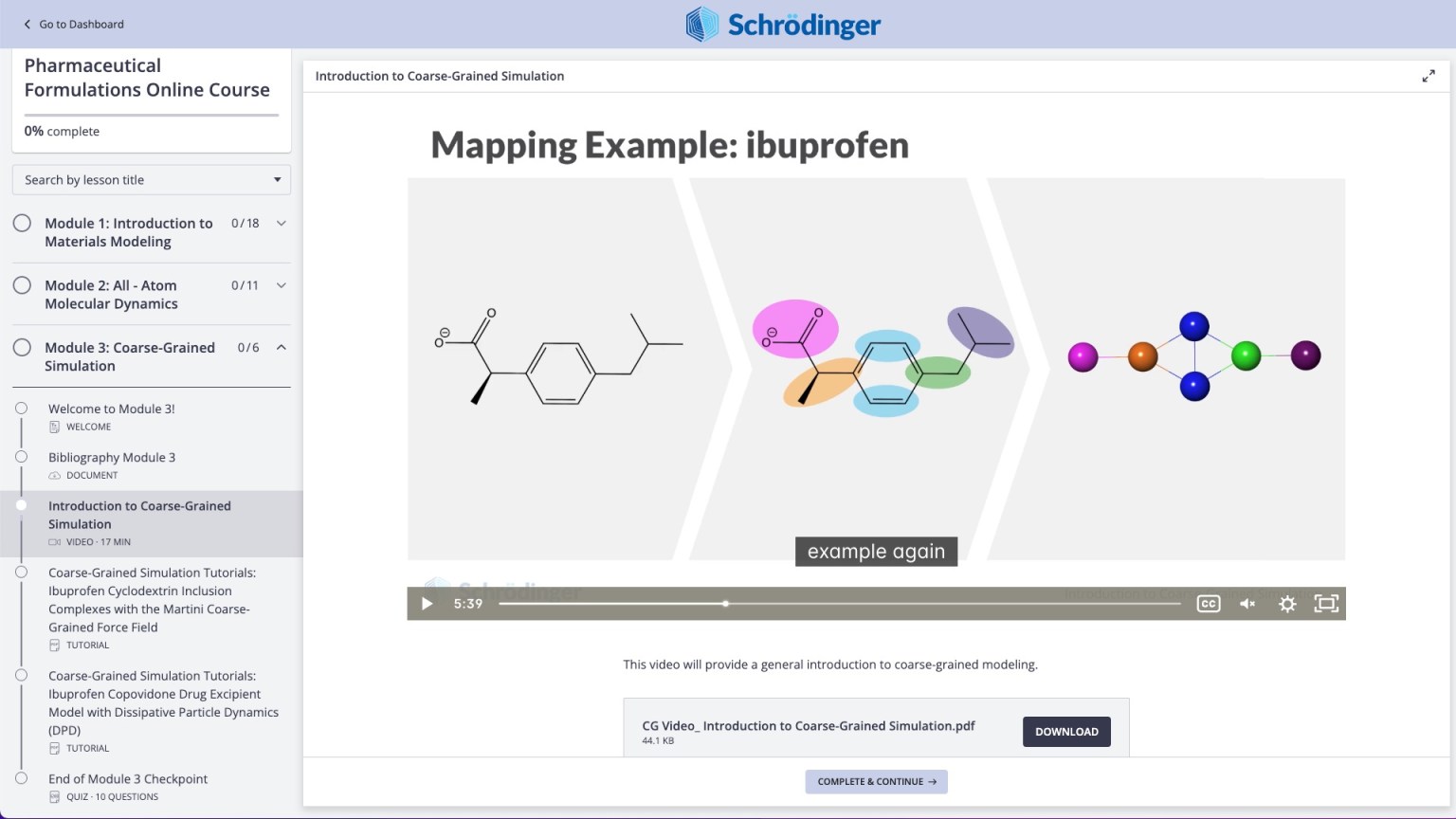
Coarse-grained modeling
Access larger length scale and longer time scales by employing coarse-grained methods to study formulations
Hands-on modeling in the web-based graphical user interface (e.g. Polymeric Materials course, Diffusion tutorial)
Videos on practical theory break down complex scientific concepts (e.g. Molecular Dynamics)
On-demand video lessons on materials modeling
Access cloud-based computing resources to perform calculations yourself
Perform case studies with expert feedback (e.g. Organic Electronic Course, Independent Case Study)
Video on practical theory break down complex scientific concepts (e.g. Machine Learning for Chemistry)
Videos on practical theory break down complex scientific concepts (e.g. Periodic Quantum Mechanics)
Videos on practical theory break down complex scientific concepts (e.g. Coarse-Graining)
Need help obtaining funding for a Schrödinger Online Course?
We proudly support the next generation of scientists and are committed to providing opportunities to those with limited resources. Learn about your funding options for our online certification courses as a student, post-doc, or industry scientist and enroll today!
“Clear instructions with a well-designed interface allowed me to run some of my own first molecular dynamics simulations. The information from the course felt much more secure than the information from YouTube because I knew it was developed by experts”
Graduate Student
“The course let me talk confidentially about molecular modeling and what it can do. For me, this was a nice experience which left me with many ideas for applying molecular modeling in the research area of our department, not only for me but also for my colleagues.”
Graduate Student
“As always, the course is very well designed. Formulation is quite outside my comfort zone in terms of theory and modeling but this course provided me with knowledge of evaluating what modeling can facilitate in the real world. Really great design and education process.”
Senior DirectorTherapeutic Protein Design
Show off your newly acquired skills with a course badge and certificate
When you complete a course with us in molecular modeling and are ready to share what you learned with your colleagues and employers, you can share your certificate and badge on your LinkedIn profile.
Frequently asked questions
How much do the online courses cost?
Pricing varies by each course and by the participant type. For students wishing to take these courses, we offer a student price of $150 for introductory courses, $305 for the Materials Science bundle, and $870 for advanced courses. For commercial participants, the course price is $575 for introductory courses and $1435 for advanced courses and bundles.
When does the course start?
The courses run on sessions, which range from 3-6 week periods during which the course and access to software are available to participants. You can find the course session and start dates on each course page.
What time are the lectures?
Once the course session begins, all lectures are asynchronous and you can view the self-paced videos, tutorials, and assignments at your convenience.
How could I pay for this course?
Interested participants can pay for the course by completing their registration and using the credit card portal for an instant sign up. Please note that a credit card is required as we do not accept debit cards. Additionally, we can provide a purchase order upon request, please email online-learning@schrodinger.com if you are interested in this option. If you have any questions regarding how to pay for the course, please visit our funding options page.
Are there any scholarship opportunities available for students?
Schrödinger is committed to supporting students with limited resources. Schrödinger’s mission is to improve human health and quality of life by transforming the way therapeutics and materials are discovered. Schrödinger proudly supports the next generation of scientists. We have created a scholarship program that is open to full-time students or post-docs to students who can demonstrate financial need, and have a statement of support from the academic advisor. Please complete the application form if you qualify for our scholarship program!
Will material still be available after a course ends?
While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course.
Do I need access to the software to be able to do the course? Do I have to purchase the software separately?
For the duration of the course, you will have access to a web-based version of Maestro, Bioluminate, Materials Science Maestro and/or LiveDesign (depending on the course). You do not have to separately purchase access to any software. While access to the software will end when the course closes, some of the material within the course (slides, papers, and tutorials) are available for download so that you can refer back to it after the course. Other materials, such as videos, quizzes, and access to the software, will only be available for the duration of the course. Please note that Schrödinger software is only to be used for course-related purposes.
Imagine having the ability to explore all of chemical space and predict the best possible molecule for a given application. The potential for breakthroughs would be limitless.
At Schrödinger, this is the vision that drives us. We’ve assembled a global team of scientists and specialists united by our mission to improve human health and quality of life by transforming the way therapeutics and materials are discovered.
With more than 30 years of continuous investment in R&D, Schrödinger has built the leading computational platform for molecular discovery.
Our own therapeutics and material science groups use the platform to advance a pipeline of proprietary and collaborative programs.
Our platform leverages the physics of molecular interactions accelerated by cutting-edge machine learning to accurately simulate and predict key properties of billions of novel molecules across vast chemical space.
The feedback we gain from our customers and internal projects helps continuously improve the platform, enhancing and expanding its predictive power and utility across applications.
Driven by the relentless pursuit of principled innovation and defined by our collaborative spirit, Schrödinger is pushing the frontier of molecular discovery.
And we believe this is just the beginning.
Join us for what’s next.
Hear from our team
At Schrödinger, we are united by a common goal — to change the world through scientific advancement. Learn more about what inspires us:
With over 800 employees across more than a dozen offices, Schrödinger’s footprint spans the globe. Our team brings diverse perspectives, experiences, and backgrounds, united by a shared purpose.
We are driven to be the world leader in transforming drug discovery and materials design by relentlessly pursuing scientific and technology breakthroughs.
We are committed to achieving the best possible outcomes for our customers, partners, patients, and other stakeholders.
We deeply value our dedicated employees and invest in their growth, development, and well-being.
We help and support each other, generously and with compassion.
We pursue a diverse, equitable, and inclusive workplace where teamwork and collaboration are valued, and open, respectful debate is welcome and encouraged.
We strive to do the right thing, applying the highest ethical standards to our work and always considering how our actions impact individuals and communities who depend on us.
Schrödinger at a glance
30+
years of innovation
~900
employees
12
offices globally
America’s 100 Most Loved Workplaces 2022: No. 1 in Pharmaceuticals and Biotech; No. 21 overall
Best-Led Companies Iist 2021
Largest Bioscience and Health Technology Companies in Oregon & S.W. Washington 2021
Largest SoftwareDevelopment Firms 2021
Innovation Awards 2021
Fastest-Growing Companies 2022
We started with a vision.
What if we could remove the boundaries of possibility when exploring chemical space?
Founded in 1990 with continuous investment in R&D, Schrödinger has pioneered a physics-based computational platform that enables the rapid and accurate discovery of high-quality, novel molecules for drug development and materials applications.
Our multidisciplinary drug discovery team deploys our platform internally to advance a portfolio of collaborative and proprietary programs to address unmet medical needs.
Dramatically improving hit rates with a modern virtual screening workflow
Scientists from Schrödinger’s Therapeutics Group leveraged a modern virtual screening workflow powered by ultra-large scale docking and absolute binding free energy calculations to achieve a double-digit hit rate for diverse protein targets.
Schrödinger developed a modern virtual screening workflow for small molecule ligands and fragments that enabled Schrödinger’s Therapeutics Group to repeatedly achieve unprecedented success in its hit discovery efforts
The modern virtual screening workflow efficiently screens ultralarge libraries of up to several billion purchasable compounds with unrivaled accuracy, through machine learning enhanced docking and absolute binding free energy calculation technologies
The workflow was successfully applied to a broad range of targets across multiple screening campaigns, for both whole ligands and fragments
Schrödinger’s Therapeutics Group used the workflow to identify multiple experimentally confirmed hits with diverse chemotypes while dramatically narrowing down the number of compounds made or purchased and assayed in the lab — frequently achieving double-digit hit rates
Background
For years, hit discovery efforts using traditional virtual screening (VS) approaches have suffered from low hit rates, typically 1-2% in Schrödinger’s experience, which means that 100 compounds would have to be synthesized and assayed for 1-2 hits to be identified. These challenges have largely been attributed to two key factors:
First, traditional VS campaigns have been limited to libraries in the hundreds of thousands to a few million in size, providing limited coverage of chemical space. This is particularly critical for difficult-to-drug targets, where the random hit rate in the library is expected to be low, hence fewer hits are expected to be recovered with smaller libraries. In recent years, the emergence of ultra-large commercial chemical libraries such as Enamine REAL and research demonstrating the value of screening large libraries has further driven the need for technologies that can efficiently screen ultra-large chemical space.1
Second, traditional VS methods have been limited by the inaccuracy of the scoring methods utilized to rank order different ligands, such as GlideScore. Given a static view of the complex geometry and an approximate treatment of desolvation, such empirical scoring functions aren’t theoretically suited to quantitatively rank compounds by affinity. Thus, while docking is a powerful technology for early enrichment, ligand docking scores are not expected to and generally do not correlate with measured potency.
As a result of these limitations, most resources spent on virtual screens using traditional methods are often wasted. A more cost-effective and efficient approach to accurately screen ultra-large libraries is required to improve the success of virtual screens and ensure it is a viable path for hit discovery.
Design Approach
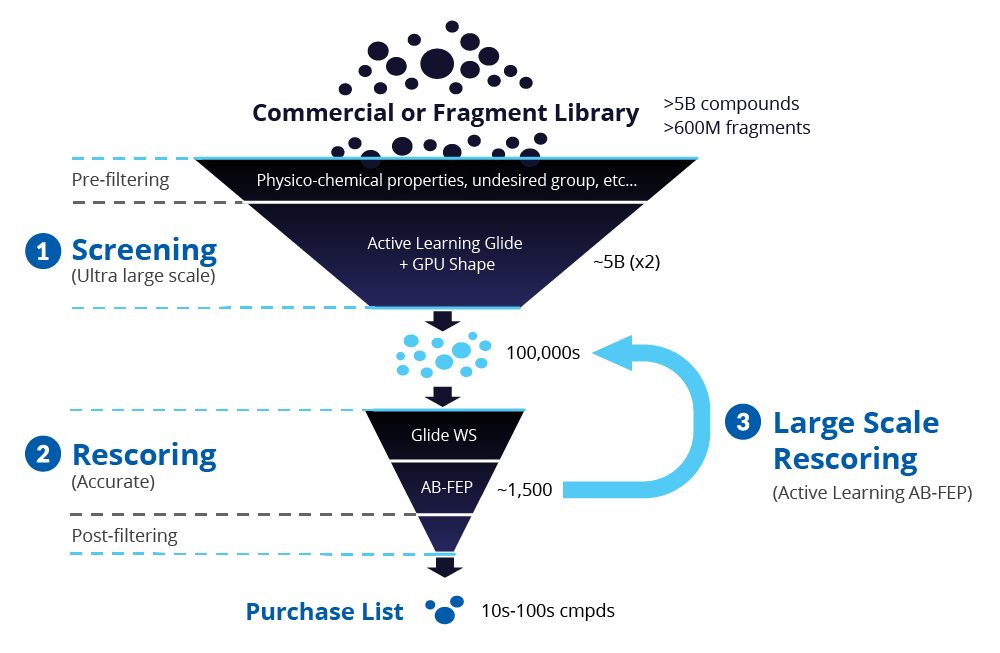
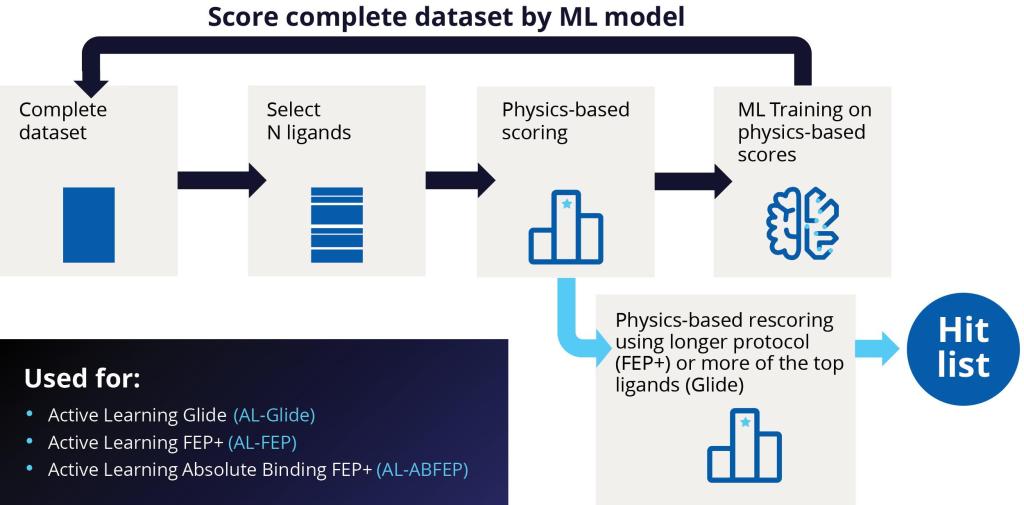
Over several years, Schrödinger’s Therapeutics Group has selected a number of challenging targets with the goal of identifying potent hit molecules. The group turned to a modern VS workflow, leveraging machine learning-guided Glide docking and highly accurate Absolute Binding FEP+ (ABFEP+) calculations, to screen and rescore ultra-large chemical libraries in a way that minimizes wet-lab costs and time while increasing the number and quality of hits available for hit-to-lead progression (Figure 1).
Figure 1: Overview of Schrödinger’s modern virtual screening workflow.
Step 1: Ultra-large scale screening for small molecule libraries
Starting with libraries on the order of several billion compounds (or libraries of up to 500 million for fragments), the team performed prefiltering based on physicochemical properties to eliminate any undesired groups. Next, they carried out a highthroughput virtual screen with Active Learning Glide (AL-Glide), in order to quickly identify the most promising compounds. Active learning is an effective supervised learning strategy that prioritizes training data for the next round of training based on a well-defined objective. AL-Glide combines machine learning (ML) with docking so that enrichment with docking can be applied to libraries of billions of compounds.2 By using this approach, only a fraction of the library is docked, reducing the computational cost significantly to a more reasonable level.
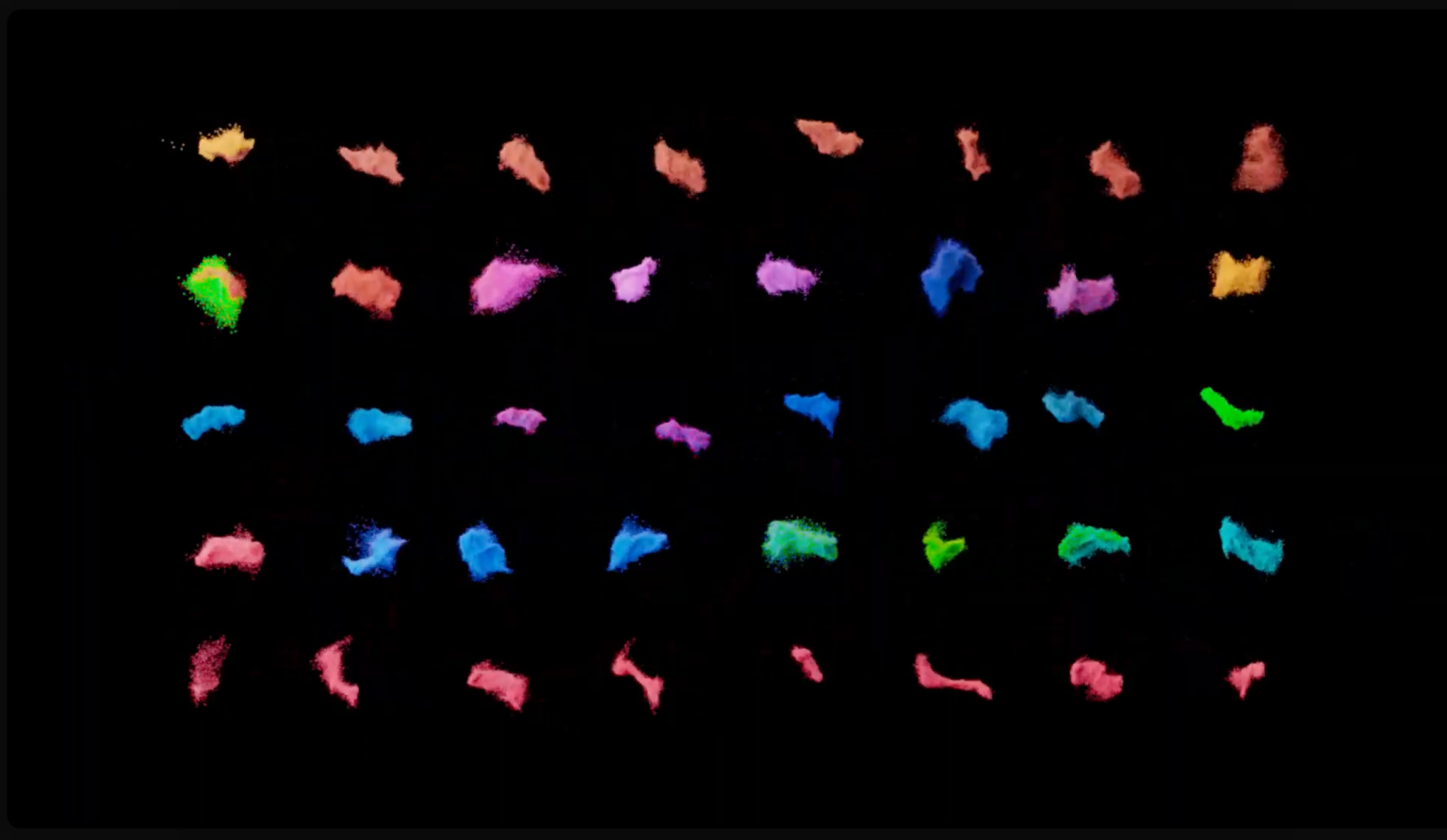
At the start of an active learning cycle, a manageable batch of compounds is selected from the complete data set of library compounds and docked. These selected compounds are then taken and added to the training set. The model is then trained on new information and continues to iterate this process as the machine learning model becomes a better and better proxy for the docking method (Figure 2).
This ML-guided docking model can evaluate compounds much more quickly than brute force docking. While the typical docking calculation with Glide might take an average of a few seconds per compound, the ML model can evaluate or make a prediction significantly faster, leading to a drastic increase in throughput. As a result, the ML-generated model is used to evaluate the entire library.
After completion of the AL-Glide screen, the team performed a full docking calculation using Glide on the best scored compounds, typically in the range of 10-100 million compounds.
Figure 2: Schematic showing an active learning workflow for hit discovery.
Step 2: Rescoring
The most promising compounds based on Glide docking scores were then selected and subjected to a rescoring step using Glide WS, a sophisticated docking program that leverages explicit water information in the binding site to enrich active molecules over Glide alone, in large part due to improved pose prediction. Glide WS helps identify better compounds to pass to ABFEP+, the next scoring step, and to provide more reliable binding poses, reducing false positives in the process.3
Compounds with the best enrichment scores from docking are selected for rigorous rescoring with ABFEP+. ABFEP+ is a protocol in Schrödinger’s FEP+ technology that allows the accurate calculation of binding free energies between the bound and unbound states of the ligand/protein complex (Figure 3).4,5 ABFEP+ has proven to reliably correlate with experimentally measured binding affinities. Unlike relative binding FEP+, ABFEP+ does not require a similar, experimentally measured reference compound as a starting point. Because ABFEP+ can evaluate and accurately score diverse chemotypes, it is a linchpin technology to discover the most potent compounds in a virtual screen campaign.
Step 3: Large-Scale Rescoring
ABFEP+ is computationally expensive when compared to Relative Binding FEP+ (RB-FEP+), requiring multiple GPUs per ligand and approximately 4x more compute time. It is generally only practical to run thousands of ABFEP+ calculations on a hit discovery campaign. In order to realize the true enrichment benefit of ABFEP+, an active learning approach is utilized to score a much larger number of compounds.
Figure 3: Thermodynamic cycle for Absolute Binding FEP+ (ABFEP+). As part of the modern virtual screening workflow on discovery programs, the accuracy provided by the rigorous calculations in ABFEP+ consistently showed very early enrichment in top actives and reduced false positives, which is critical when screening ultra-large libraries. * Compounds with molecular masses between 100 and 250 Da
Applying the modern virtual screening workflow to fragments
Experimental fragment screening has led to multiple FDA-approved drugs and clinical candidates. By adapting this modern workflow to fragment screening, Schrödinger’s Therapeutics Group has successfully scaled up screening to millions of fragments, as compared to 3k to 30k fragments screened by traditional HTS.
The in silico approach addresses a fundamental limitation of experimental fragment screening — the fragments need to be soluble enough to be assayed at high concentrations (100 μM to mM) against various targets. However, estimating the potency rigorously in silico is not limited by solubility, so the potency of the fragments can be assessed and subsequently pursued if they are predicted to be soluble enough given their estimated potency. The binding potency to the specific target is computed using active learning ABFEP+. Priority fragments are finally evaluated for solubility in silico at predicted potency using Solubility FEP+.6 In essence, the approach achieves scale by inverting the problem of potency and solubility, enabling the discovery of highly potent and ligand efficient fragments that would not exist in experimental fragment libraries.
To date, the team has completed a total of nine large fragment-based* virtual screens on multiple challenging targets, including one with a homology model. All nine screens yielded multiple potent ligand efficient hits, ranging from low nM to 30 μM in potency and double-digit hit rates.
Impact of Schrödinger’s modern VS workflow on hit rates across multiple projects and targets
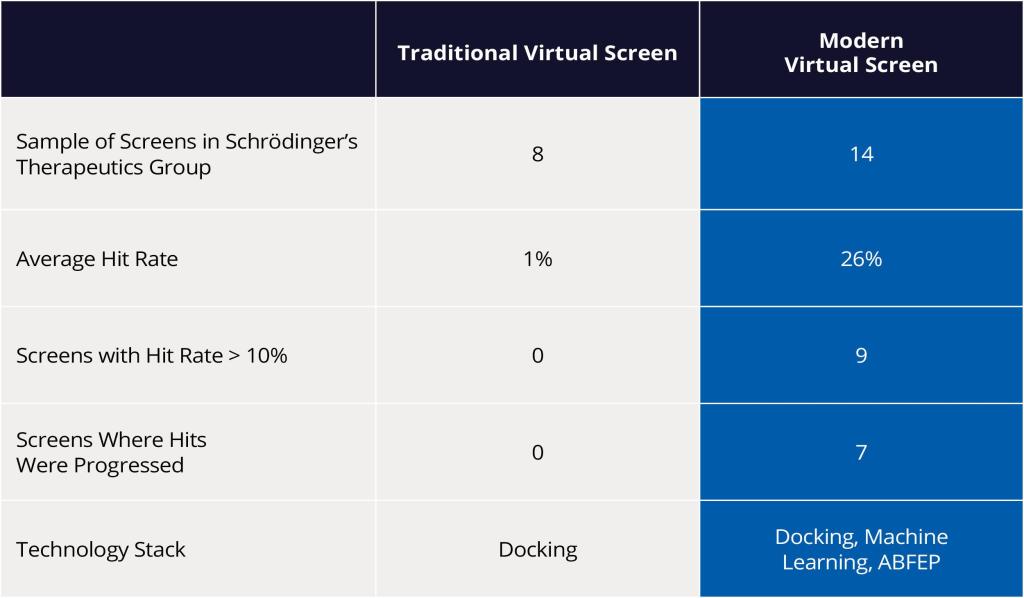
Using these modern VS approaches, scientists at Schrödinger were able to demonstrate a drastic improvement in hit rates compared to traditional screens. As a result, several diverse hit compounds with high predicted binding affinity were identified, acquired, experimentally tested and confirmed as hits — resulting in an impressive doubledigit percentage hit rate (Figure 4).
Figure 4: Impact of transitioning from traditional to modern virtual screening on hit rate.
Conclusion
In silico hit identification has long relied on smaller scale libraries and lower accuracy methods to screen the chemical space, resulting in low hit rates and largely wasted wet lab resources.
By transitioning to a modern workflow that leverages rigorous physics-based methods, including absolute binding FEP+ combined with machine learning, Schrödinger’s Therapeutics Group has been able to successfully apply the workflow to a range of diverse targets across several projects and achieve a reproducible doubledigit hit rate. In the process, the team dramatically reduced the number of compounds synthesized and tested to reach the project’s lead candidate, reducing overall costs and project timelines.
This strategy empowers drug discovery teams by enabling them to explore the fast-growing ultra-large chemical libraries. It allows efficient navigation through the vast maze of chemical space, significantly improving the odds of identifying multiple hits with better properties and selectivity. Moreover, it accelerates the drug development process, leading to the faster discovery of higher-quality, novel drug candidates.
Enabling digital technologies to drive discovery programs
FEP+
Digital assay for predicting protein-ligand binding across broad chemical space at an accuracy matching experimental methods.
Ultra-large library docking for discovering new chemotypes. Lyu J, et al.
Nature. 2019 Feb; 566(7743): 224–229.
Efficient exploration of chemical space with docking and deep learning. Yang Y, te al.
J. Chem. Theory Comput. 2021, 17, 11, 7106–7119.
WScore: A flexible and accurate treatment of explicit water molecules in ligand−receptor docking.
Murphy RB, et al. J. Med. Chem. 2016, 59, 4364−4384.
Enhancing hit discovery in virtual screening through accurate calculation of absolute protein-ligand binding free energies.
Chen W, et al. J. Chem. Inf. Model. 2023, 63, 10, 3171–3185.
Accurate calculation of the absolute free energy of binding for drug molecules.
Aldeghi M, et al. Chem. Sci. 2016;7:207–218.
Novel physics-based ensemble modeling approach that utilizes 3D molecular conformation and packing to access aqueous thermodynamic solubility: A case study of orally available bromodomain and extraterminal domain inhibitor lead optimization series.
Hong RS, et al. J. Chem. Inf. Model. 2021, 61, 3, 1412–1426.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Designing the next generation of materials starts at the atomic scale.
Combine rigorous quantum mechanics and molecular dynamics with predictive AI to explore vast chemical space, predict material properties with high fidelity, and de-risk experimental R&D.
Explore vast chemical space with digital chemistry
Get more from your ideas by harnessing the power of large-scale chemical exploration and highly accurate, in silico property predictions.
“Digital chemistry can expand the pool of hypotheses we can test. Ultimately we can unleash the power of our creativity.”
Adam LevinsonDirector, Therapeutics Group
Solutions for small molecule drug discovery
Structure Prediction & Target Enablement
01
Unlock your protein target for high-precision, structure-based design
Comprehensive suite of solutions for protein model refinement, ligand placement, and binding site analysis allows you to unlock a broader range of targets for structure-based design.
Maximize your chances of finding diverse, high-quality hits
Diverse technologies for structure-based and ligand-based screening that enable efficient, large-scale chemical exploration and highly accurate rescoring.
Optimize your drug formulation processes with structure-based insights
Computational solutions for advancing small molecule formulation, from crystalline or amorphous forms to selection of materials and excipients for processing, stability, and delivery.
This two-day, in-person user group meeting (UGM) event will bring together scientists and industry professionals to exchange ideas, explore new approaches, and connect with peers across the industry.
FEP+ is a widely adopted technology for accelerating small molecule drug discovery programs, with application in hit discovery, hit-to-lead, and lead optimization. The accuracy and utility of FEP+ as a computational assay for the prediction of binding energies is validated extensively, with predictions falling within 1.0 kcal/mol of experimental values on average.1,2 With accuracy approaching lab experiment, FEP+ serves as a digital binding affinity assay for both on- and off-targets which enables drug discovery teams to efficiently optimize molecular profiles and pursue novel chemistry with confidence.
Given the computational cost and complexity of running free energy calculations, adoption is largely driven by expert computational chemists who then manually share results with the project team. To amplify the impact of these methods and improve the agency of individual team members, they should be accessible directly to medicinal chemists and be able to be run at a scale comparable with traditional virtual screening methods.
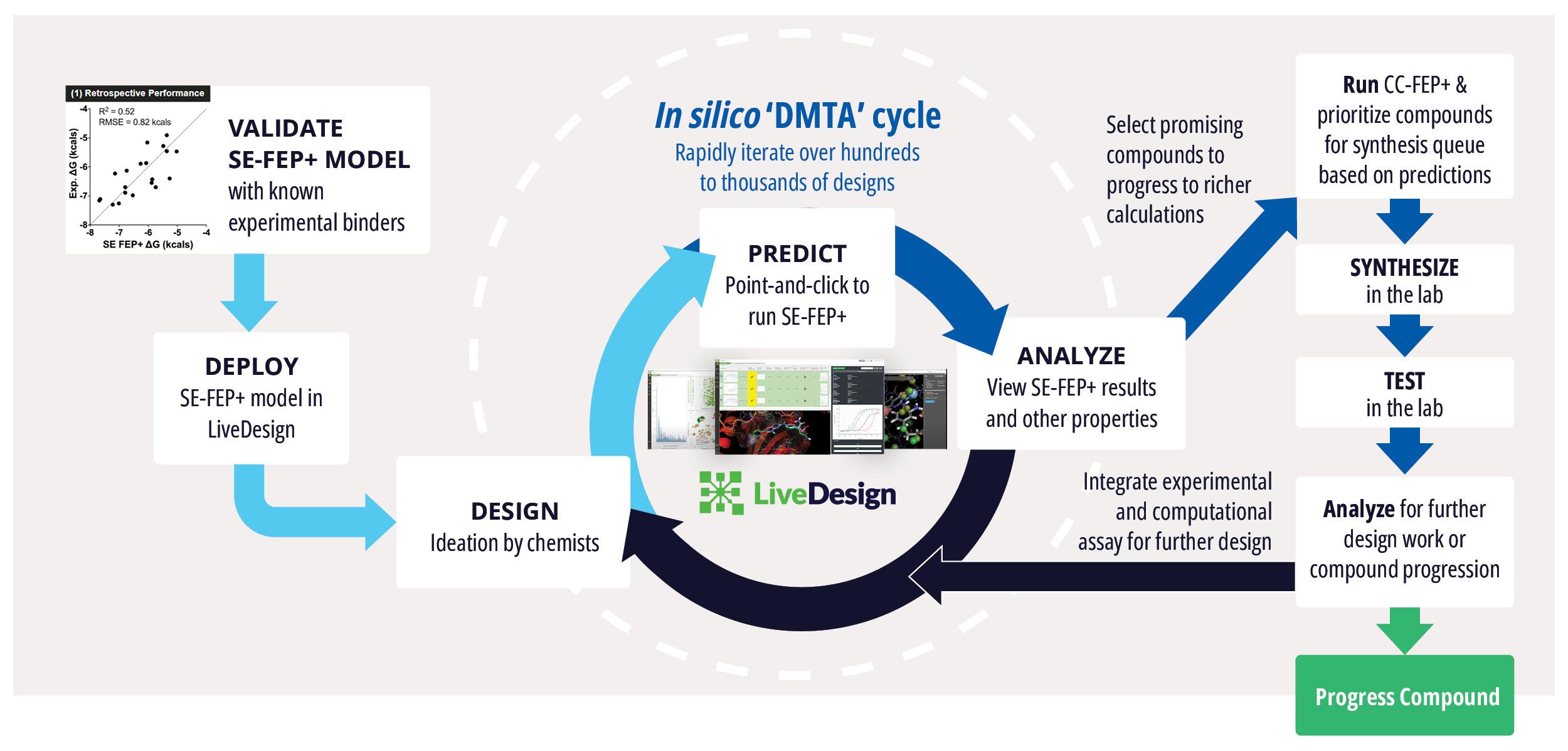
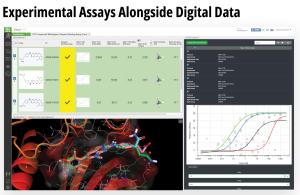
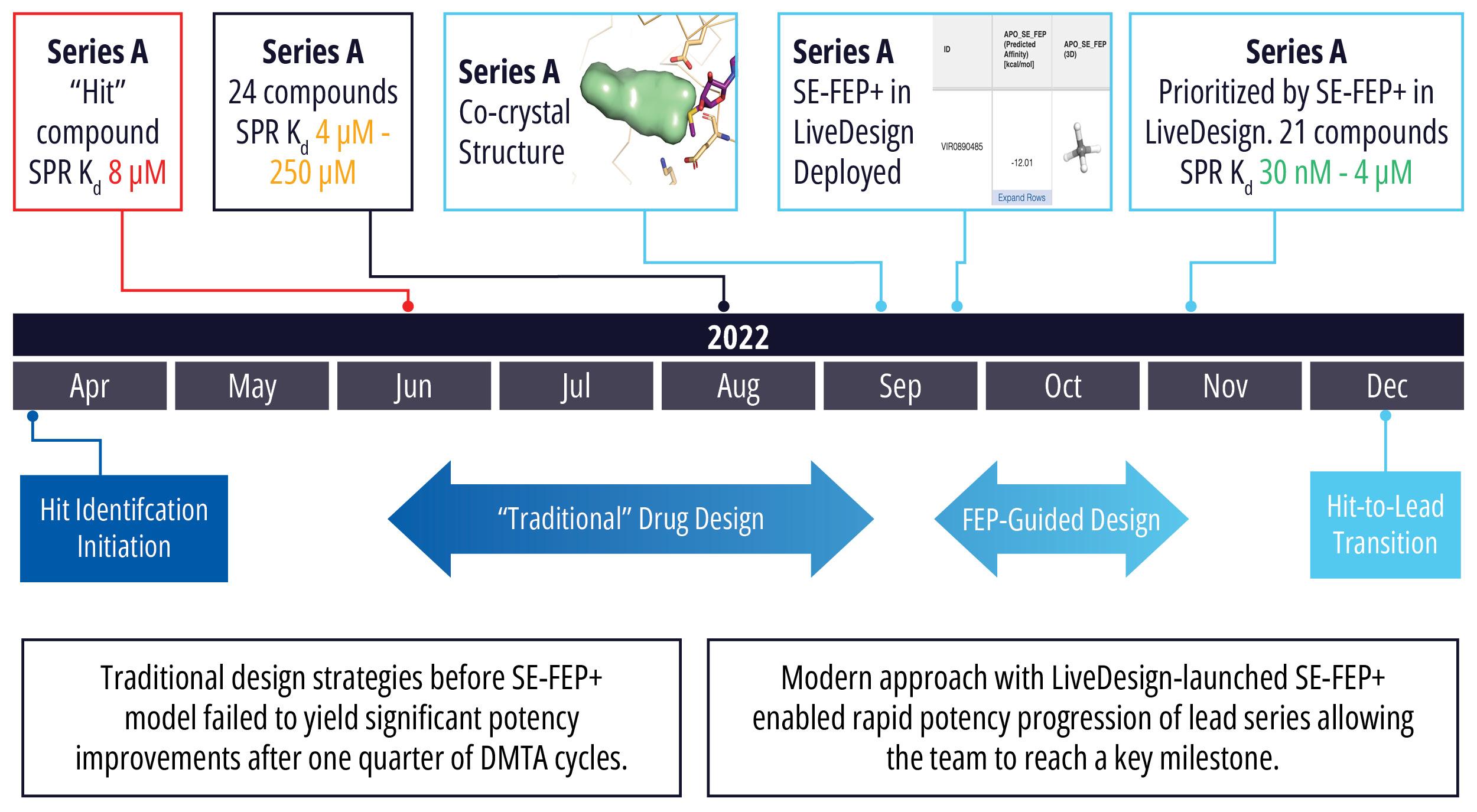
Toward these goals, developments in expanding the accessibility of free energy calculations — in particular single-edge FEP+ (SE-FEP+) — enable a new paradigm in structure-based molecular design. SE-FEP+ simulations can be run in a fraction of the time of full cycle-closure FEP+ (CC-FEP+) simulations and can be conceptualized as ‘push-button’ for execution. This makes it easily deployable in LiveDesign, a cloud-native, collaborative enterprise design platform. This workflow enables all project team members to integrate FEP+ within their design workflows, resulting in a highly interactive and fully in silico design-make-test-analyze (DMTA) cycle where chemists are empowered to digitally test hypotheses and iteratively improve designs prior to compound synthesis (Figure 1).
In one such oncology program within Schrödinger’s Therapeutics Group, the project team was able to improve compound potency over 100-fold by running many iterative in silico DMTA cycles using FEP+ in LiveDesign over the course of a four week period.
Figure 1: Process for deploying and running in silico DMTA Cycles via SE-FEP+ and its potential for tight integration with traditional experimental DMTA cycles.
Single-Edge FEP+:
Accurate prediction of relative binding affinities in a fraction of the time
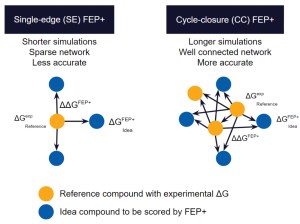
As compared to docking or MM-GBSA calculations, full cycle-closure FEP+ (CC-FEP+) simulations enable highly accurate rank-ordering of compounds, but are compute intensive and complex to set up, reducing their ability to be set up and executed by non-expert molecular modelers.
Single-edge FEP+ (SE-FEP+) simulations have been shown to maintain high predictive accuracy but can often be run ~10x faster than CC-FEP+ simulations in an automated fashion to triage new designs efficiently. Once the SE-FEP+ model is validated, it can be used in a push-button fashion to test new designs that share a common scaffold (i.e., common core or common R-group) with the reference compound and enable efficient scoring of large numbers of idea compounds (Figure 2).
When deployed via a collaborative platform such as LiveDesign, SE-FEP+ gives project teams the ability to run rapid design cycles and prioritize ideas with confidence. The most promising design ideas can then be run through full CC-FEP+ calculations to obtain the most robust potency predictions for informing synthesis queue decisions. If a compound falls outside the criteria established by the computational chemist for running the SE-FEP+ models, that molecule can be added to a queue for running full CC-FEP+ in a more manual setup by a computational chemist.
Figure 2: Schematic representation of Single-Edge FEP+ and Cycle-Closure FEP+. During single-edge FEP+, idea compounds are evaluated against a single reference compound and usually for a fraction of the simulation length compared to full CC-FEP+ calculations, resulting in a faster albeit less accurate calculation. During cycle-closure FEP+, design ideas are usually evaluated against multiple reference compounds in an interconnected map with longer simulation lengths for the highest accuracy results.
A complete digital molecular design lab
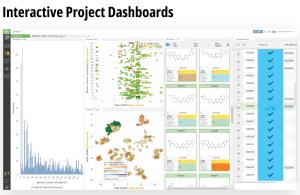
LiveDesign is a flexible, cloud-native collaborative working environment for entire discovery teams. The platform democratizes digital design processes and enables more productive design cycles by harnessing the power of medicinal chemistry strategies, advanced cheminformatics, cutting-edge computational chemistry workflows, virtual ‘design and test’ technologies, and centralized access to live project data — all in a single interface.
The platform empowers creativity by democratizing access to powerful predictive modeling workflows, such as push-button free energy calculations, and allows teams to efficiently capture and progress their most promising design ideas. LiveDesign also centralizes team collaboration and decisionmaking — enabling crowdsourcing of design ideas, interactive team-based design, and easy sharing of data with internal and external partners.
Case Study:
Oncology Program SCH-01
In an early stage oncology drug discovery program driven by Schrödinger’s Therapeutics Group, the project team faced a design challenge to find and optimize an initial hit series. Of the 200 compounds synthesized in the program to date, the most potent molecule had a modest binding affinity of 2.7 μM, which failed to meet the criteria for the hit-to-lead transition. Traditional design strategies had failed to yield significant potency improvements after several months of DMTA cycles, even in the most promising series.
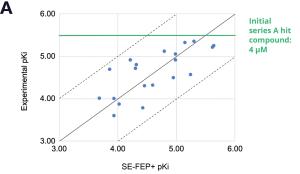
With a new cocrystal structure for this promising series in hand, the team turned to FEP+ to address the potency challenge. An SE-FEP+ model was developed and validated retrospectively using known experimental binding affinities. The SE-FEP+ model was then deployed in LiveDesign, allowing medicinal chemists to design new ideas and independently run SE-FEP+ calculations with the click of a button.
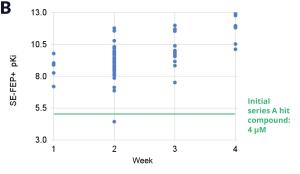
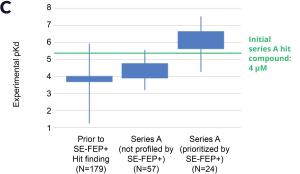
Using this validated model, the project team iteratively designed molecules and digitally assessed their ability to improve potency from the current best Series A potency of 4 μM. During one particular four week period, approximately 400 designs were profiled with medicinal chemistry team members contributing and running SE-FEP+ in LiveDesign for ~65% of the designs. No new compounds were experimentally synthesized and tested in this month, but the team was able to significantly optimize compound potency in silico as predicted by FEP+. Each week, the predicted potency of the designs made by the chemistry team improved until the top scoring compounds were then prioritized for synthesis and experimental profiling (Figure 3).
By prioritizing 24 compounds for synthesis based on SE-FEP+ predictions in LiveDesign over this one month period, the project team dramatically enriched the synthesis queue with potent designs. In fact, 21 compounds exhibited improved measured binding affinities of less than 4 μM (Figure 4). Based on this significant progress, the team moved the program forward from the hit identification stage to the hit-tolead stage in a highly accelerated time frame.
Figure 3: Rapid potency improvements from SE-FEP+ in LiveDesign over a four week period. A) Initial retrospective validation of single edge FEP+ model performance vs experiment (known binders) B) Predicted binding affinity by week for new designs profiled directly in LiveDesign over a four week period demonstrates rapid improvement as the team iterated on idea compounds in silico. C) Confirmed experimental binding demonstrates over 100 fold improvement from compounds prioritized by the SE-FEP+ in LiveDesign workflow compared to those designed prior to SE-FEP+.
Figure 4: Impact of SE-FEP+ in LD on program hit identification timeline
Conclusion:
Deployment of SE-FEP+ in LiveDesign presents a new paradigm for computationally-guided design by democratizing access to powerful, push-button FEP+ calculations to teams of medicinal chemists. It enables chemists to think broadly and creatively when designing ideas by lowering the barrier to in silico testing of ideas before synthesis. Additionally, the workflow enables expert computational chemists to create guardrails around the use of FEP+ by chemistry teams, and focus time and effort on the more complicated full-cycle challenges to the highly triaged compounds.
This workflow can assist in rapid potency progression, optimization of selectivity against known off-targets, and maintenance of potency while optimizing other physicochemical properties. This allows project teams to quickly reach key milestones and design the best possible molecules to meet the target product profile while dramatically reducing the number of compounds synthesized and tested.
How it works
Succcesful deployment of SE-FEP+ in LiveDesign follows these key steps:
A scientist trained in FEP+ creates and validates an FEP+ model on a specific protein-ligand series of interest.
A model execution strategy is determined around permitted structural modifications, licenses, and available compute hardware as these calculations are computationally significant.
The FEP+ model is uploaded into LiveDesign and shared via a user-input constrained model.
In LiveDesign, collaborators design structural modifications to the reference molecule and run SE-FEP+ calculations through the push of a button.
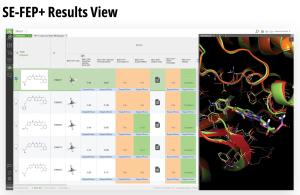
Results are provided directly in LiveDesign, including the corresponding predicted changes in free energy for each design idea, simulation statistics, and the 3D pose of the ligand in the protein.
Teams can continue to ideate and explore modifications to identify promising compounds. The most promising compounds are then progressed and further scored with CC-FEP+ for the highest accuracy binding affinity prediction.
Best Practices
Key points to consider before deploying SE-FEP+ in LiveDesign:
Ensure your SE-FEP+ model is well-validated using known experimental binding affinities.
Educate all project team members on accurate interpretation of model predictions and to build appropriate confidence in the SE FEP+ model. The Schrödinger Online Course, “Free Energy Calculations for Drug Design”, is a useful resource to learn more about FEP+, including a hands-on case study.
Develop template reports in LiveDesign that include automated safeguards around what structural modifications can be run successfully using the SE-FEP+ models.
Ensure sufficient cluster compute resources are secured to run SE–FEP+ simulations efficiently based on anticipated throughput.
Schrödinger is happy to partner with users to share best practices and provide guidance on optimizing FEP+ workflows for discovery programs. Reach out to your local Schrödinger team for assistance.
References
Impacting Drug Discovery Projects with Large-Scale Enumerations, Machine Learning Strategies, and Free-Energy Predictions
Knight et al. ACS Symposium Series, 1397, 2021, 205-226
Advancing Drug Discovery Through Enhanced Free Energy Calculations
Abel et al. Acc. Chem. Res. 2017, 50, 7, 1625–1632
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Schrödinger デジタル創薬セミナー: Into the Clinic ~計算化学がもたらす創薬プロセスの変貌~
Share
Speakers
Goran Krilov
Senior Director, Schrodinger Therapeutics Group
Abstract
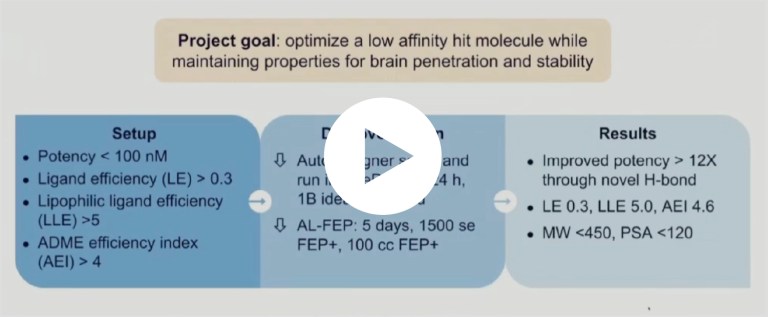
Mucosa-associated Lymphoid Tissue Lymphoma Translocation Protein 1 (MALT1) is a genetically validated target for the treatment of diseases associated with lymphocyte regulation. Unlike first generation inhibitors, centered on large peptidomimetics targeting the protease domain, second generation inhibitors targeting an allosteric region at the interface of the caspase-like and Ig3 domains are much more promising. Still, significant challenges remain in optimizing properties such as permeability, efflux, and solubility, while maintaining on-target potency. Harnessing the full potential of the Schrödinger platform which combines rigorous physics-based modeling with machine learning (ML), predictive ADMET models, and data analytics to search and triage a chemical space, we were able to rapidly identify multiple novel potent series. Subsequent in-silico multi parameter optimization (MPO) campaign quickly identified SGR-1505, a potential best-in-class MALT1 inhibitor with balanced properties and on-target activity, within 10 months of the start of the project and having synthesized only 129 compounds. SGR-1505 demonstrates strong positive effects in patient-derived B-cell tumor models both as a single agent as well as in combination with existing standard of care, and is currently progressing through Phase I clinical trials.
Performed rapid in silico design cycles using a collaborative platform and a large-scale de novo design workflow
Optimized potency and selectivity with relative binding FEP+ and protein FEP+
Resulted in a development candidate currently in preclinical development
Target
Wee1, Ser/Thr kinase
Program Type
Schrödinger proprietary program, small molecule
Indication
Solid tumors
Stage
Phase 1 clinical trial
“This program demonstrates the first prospective application of protein FEP+ to model broad kinome selectivity, which not only enabled the Wee1 team to rapidly identify a few highly selective chemotypes and rescue a program that had hit a major roadblock, but it also added a powerful tool for the broader drug discovery community to tackle selectivity challenges more efficiently.”
Jiashi Wang
Senior Director, Medicinal Chemistry
Schrödinger Therapeutics Group
Design challenge
Inhibition of Wee1, a serine/threonine protein kinase which serves as the gatekeeper of the G2-M cell-cycle checkpoint, forces cells into unscheduled mitosis and culminates in cell death. Given its critical role in DNA repair, Wee1 is an attractive target for oncology drug development. Clinical trial data for the Wee1 inhibitor adavosertib (AZD-1775) validated the potential of targeting Wee1 by revealing strong anti-cancer activity in solid tumors.1-11 However, it was suspected that AZD-1775 had potential liabilities due to inhibition of several other kinases, including the PLK family, as well as time-dependent inhibition of CYP3A4.
The aim of this program, driven by Schrödinger Therapeutics Group, was to develop a best-in-class, highly selective Wee1 inhibitor by leveraging rigorous physics-based modeling approaches to address off-target liabilities with high precision.
Efficient generation of a selective and potent lead series with a rigorous FEP-based workflow
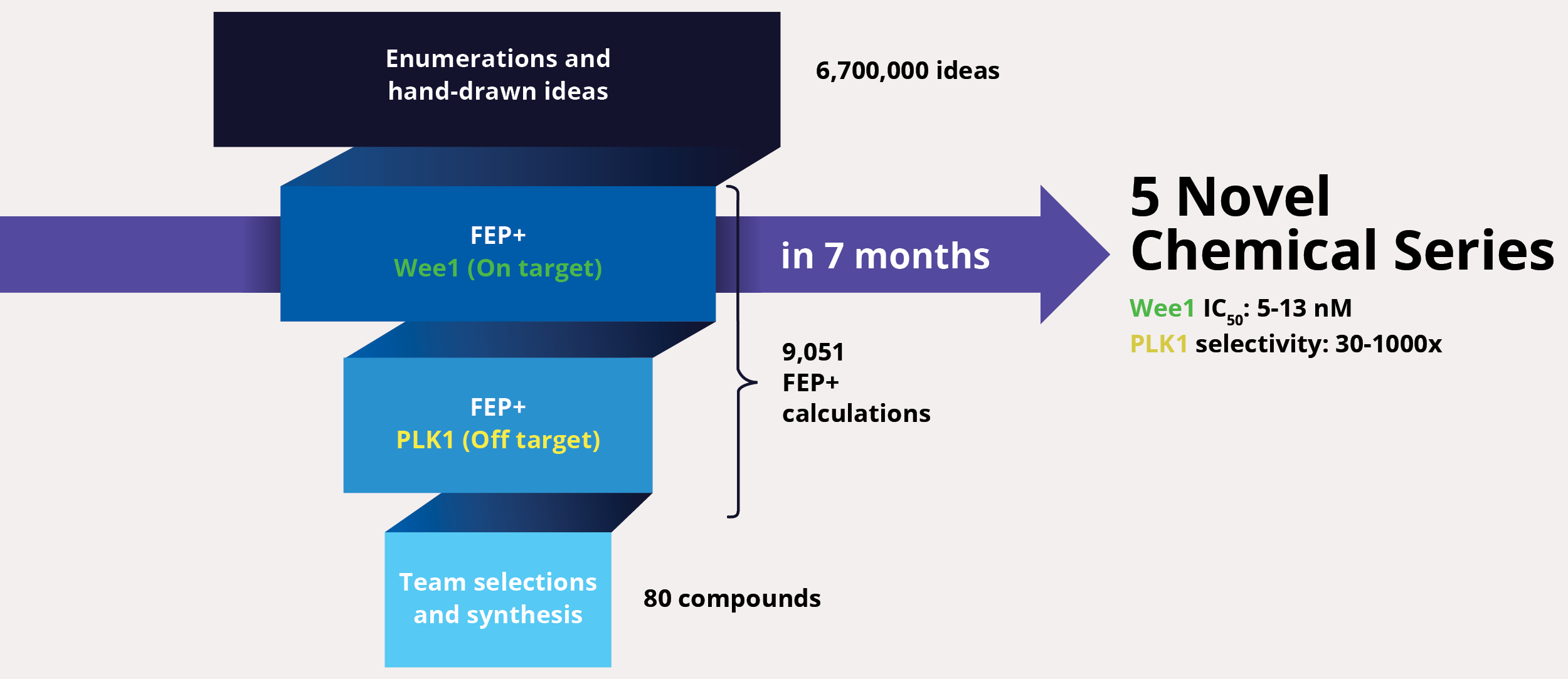
At project onset, the team’s goal was to identify one or multiple novel lead series with improved selectivity for Wee1 as compared to polo-like kinase 1 (PLK1). The team enumerated over six million ideas and hand-drawn designs, then triaged them using an automated chemistry-based filtering workflow, structure-based screening (docking), and ultimately rigorous calculation of relative binding free energies via free energy perturbations (FEP+, De Novo Design Workflow) (Figure 1). The accuracy and utility of FEP+ as a computational binding affinity assay has been validated extensively, generating predictions within 1.0 kcal/mol of experimental values on average.12 By combining FEP+ with high performance cloud computing and machine learning (Active Learning FEP+), over 9,000 FEP+ calculations were performed to evaluate the affinity of the most promising designs for inhibiting Wee1 and ensuring selectivity over PLK1.
Design ideas were then evaluated by the project team in LiveDesign, Schrödinger’s cloud-based enterprise informatics platform. These workflows informed the selection of less than 100 compounds for synthesis and testing, resulting in the identification of multiple novel chemotypes with nanomolar affinity and enhanced selectivity (up to 1000x) for Wee1 over PLK1 within seven months from project start.13
Figure 1: FEP+ workflow to identify potent and selective Wee1 inhibitors, and enhance selectivity for Wee1 over PLK1.
Addressing additional off-target liabilities with protein FEP+
Although the team successfully improved selectivity against PLK1 in the first phase of the project, subsequent kinase panel screening revealed a significant number of unanticipated off-target kinase liabilities. In lieu of starting over or scrapping the program entirely, the team explored a complementary strategy to improve selectivity of the chemotypes identified in the first phase. A major driver of selectivity seemed to be a specific residue in the binding site. The team utilized protein FEP+ calculations, a protocol within FEP+, to assess the impact of single point-mutations at that location on the ligand affinity and to infer selectivity across a large diversity of kinases without the need to profile each kinase separately. In this new workflow, 6,700 new designs were profiled with ligand FEP+ for predicting potency against Wee1 and with protein FEP+ for predicting broad kinome selectivity. During this three month modeling campaign, the ligand FEP+ and protein FEP+ strategy identified 42 promising molecules for synthesis and 22 of these molecules exhibited low nanomolar to picomolar measured potencies against Wee1 with substantially reduced selectivity liabilities (Figure 2).14
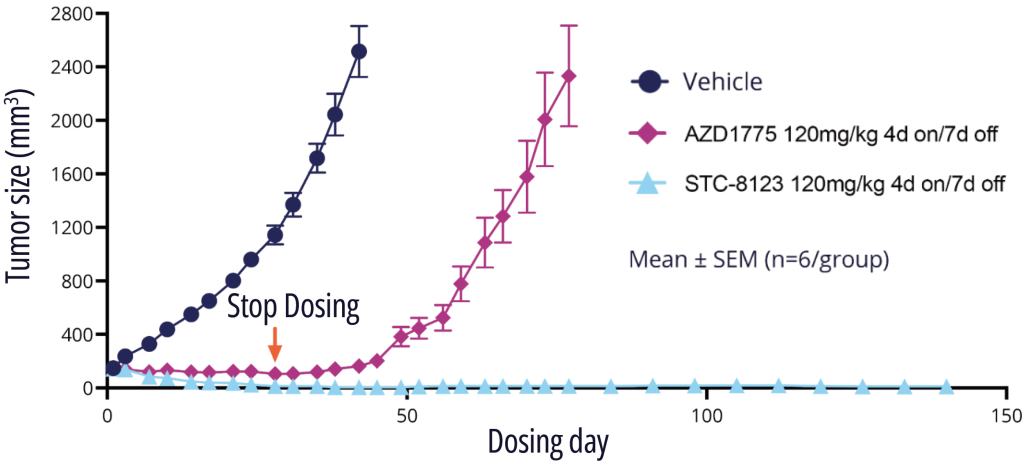
One exquisitely selective molecule, STC-8123, was used as a proof-of-concept compound to demonstrate that highly selective Wee1 inhibitors retain profound in vivo efficacy. Indeed, in an A427 mouse model with intermittent dosing, STC-8123 demonstrated rapid and more sustainable inhibition of tumor growth compared with AZD-1775 (Figure 3).14
Figure 2: Kinome selectivity profiling using scanMAX Kinase Assay Panel confirms that the FEP+/Protein FEP+ workflow enabled the rapid optimization of non-selective hits into multiple series with gene-family wide selectivity — as exemplified by STC-8123.
Figure 3: No tumor regrowth observed after STC-8123 high dose treatment stopped in A427 (non-small cell lung adenocarcinoma) tumor model.14
Optimizing DMPK and ADME properties with machine learning and quantum mechanics strategies
With a promising chemical series in hand, the team began the process of optimizing DMPK and ADME properties. As experimental data accumulated for the lead series, the team trained project-specific machine learning (ML) models to predict time-dependent inhibition of CYP3A4 profiles as well as a variety of ADME properties (DeepAutoQSAR). In addition, quantum mechanical (QM) calculations were developed for modeling compound reactivity with the CYP3A4 heme, a potential target for drug-drug interaction liabilities (Jaguar). During late-stage lead optimization, this breadth of ML models and QM physics-based calculations enabled prospective multiparameter optimization to readily narrow down the chemical space that was sent for synthesis and testing.
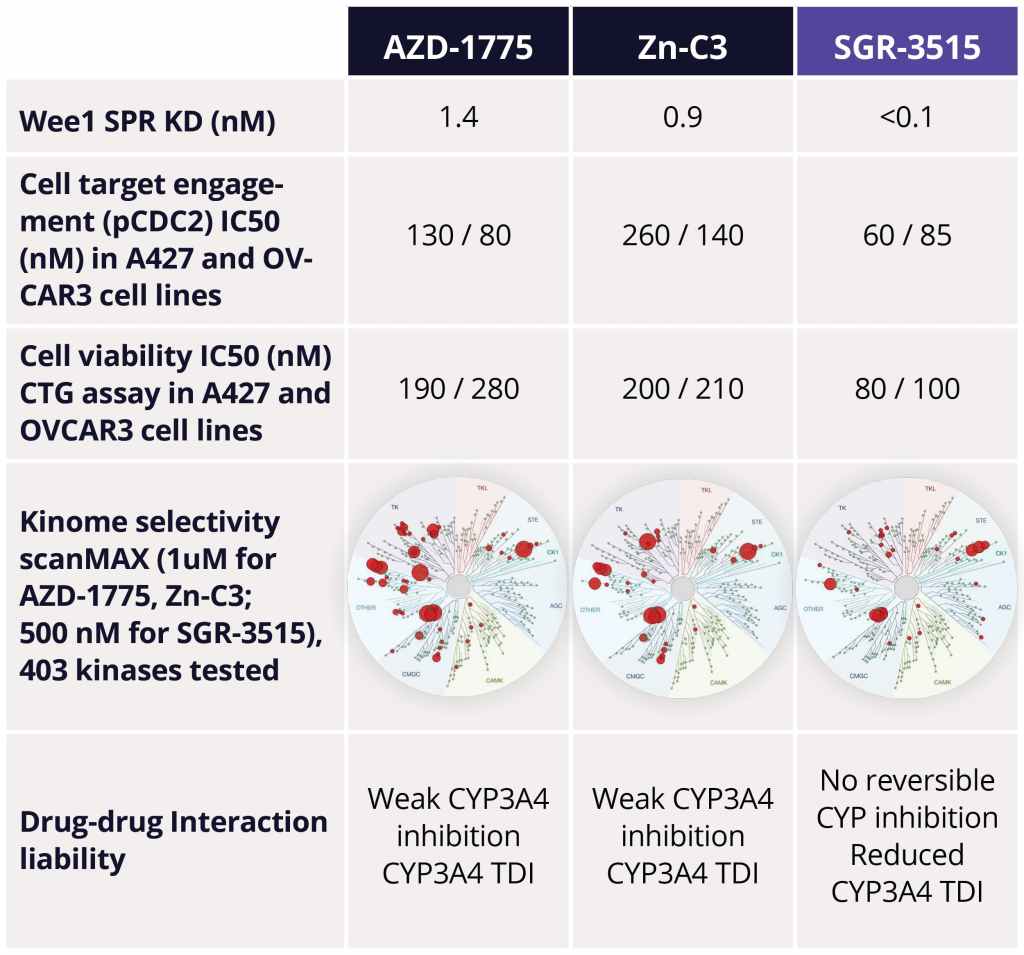
The team identified several advanced leads from among the ~300 compounds synthesized in the series. Upon in-depth profiling of advanced leads and careful consideration of the properties that a best-in-class, next generation Wee1 inhibitor should have, SGR-3515 was nominated as the development candidate. SGR-3515 is an exquisitely selective, potent molecule that is structurally-differentiated from competitors’ molecules, has a differentiated ADME profile, and achieves superior in vivo efficacy (Figure 4).
Figure 4: Comparison of SGR-3515 with competitors’ Wee1 inhibitors.15 SGR-3515 shows superior binding affinity and potency in cellular assays in A427 and OVCAR3 (high-grade serous ovarian adenocarcinoma) cell lines, demonstrates excellent broad kinome selectivity, and reduces drug-drug interaction potential due to decreased time-dependent inhibition (TDI) of CYP3A4. All competitor data was internally generated by contract research organizations.
Enabling digital technologies to drive discovery programs
FEP+
Elucidation of ligand binding preferences for large families of off-targets with protein FEP+ and on-target potency with FEP+
Cancer Res (2019) 79 (13_Supplement): CT02. 12.J Clin Oncol 2021 Nov 20;39(33).
Advancing drug discovery through enhanced free energy calculations. Abel et al.
Acc. Chem. Res. 2017, 50, 7, 1625–1632.
De-risking off-target liabilities with protein free energy methods.
Knight et al. ACS 2022.
Discovery of potent, selective, and orally available WEE1 inhibitors that demonstrate increased DNA damage and mitosis in tumor cells leading to tumor regression in vivo.
Sun et al. AACR 2022.
Into the clinic: Transforming the drug discovery process with digital chemistry.
Davis et al. Lab of the Future 2023.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.



 Blog
Blog
 Blog
Blog





 Validate protein models without experimental structures or from low resolution structures using IFD-MD with FEP+
Validate protein models without experimental structures or from low resolution structures using IFD-MD with FEP+ 











































What our alumni say