Hit to development candidate in 10 months: Rapid discovery of SGR- 1505, a novel, potent MALT1 inhibitor
Digital chemistry platform provides scale and accuracy to drive high precision molecular design
compounds computationally evaluated
total compounds synthesized in lead series
To discovery of development candidate
MALT1, protease
Schrödinger proprietary program, small molecule
Relapsed or refractory B-cell lymphoma, chronic lymphocytic leukemia
Phase 1 clinical trial
“The ability to leverage the computational platform to rapidly identify not just one, but several novel, highly potent series with well-balanced properties is unique in my many years experience in industry.”
Zhe Nie
Project Lead, Executive Director, Medicinal Chemistry,
Schrödinger Therapeutics Group
Design challenge
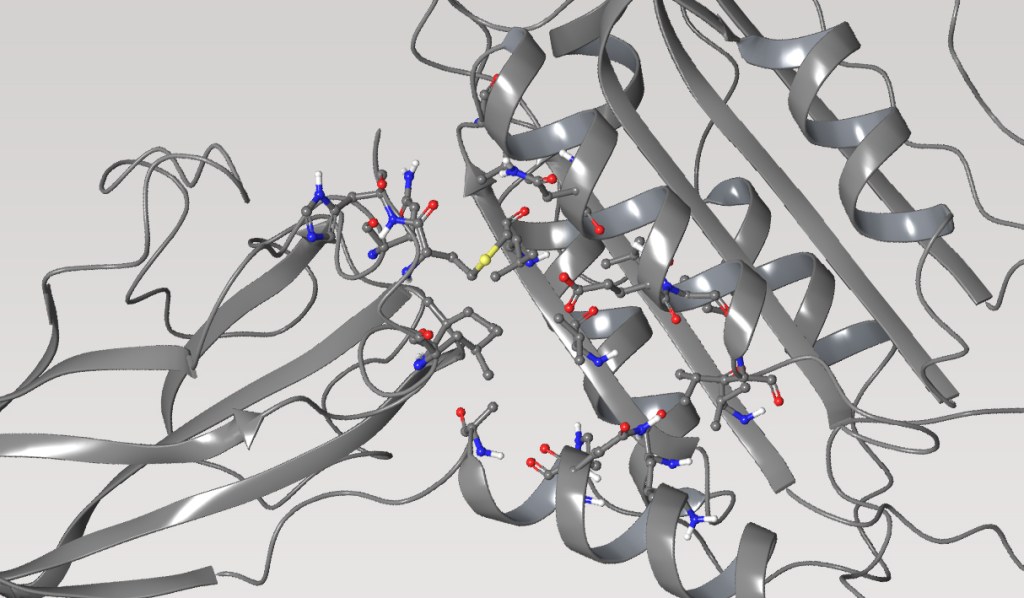
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) is a genetically validated target for the treatment of diseases associated with lymphocyte regulation. MALT1 consists of three domains: a paracaspase protease domain, an Ig3 domain, and a linking helix. First generation MALT1 inhibitors consisted of large peptidomimetics targeting the protease domain; due to their poor drug-like properties, none made it into the clinic. Second generation MALT1 inhibitors targeting an allosteric region at the interface of the caspase-like and Ig3 domains have been more successful, resulting in a clinical stage compound.
Significant challenges exist in optimizing the properties of second generation MALT1 inhibitors, specifically permeability, efflux, and solubility, while maintaining on-target potency. The aim of this program was to discover a potent inhibitor with good overall drug-like properties to support combinations with standard of care agents for treatment of relapsed or refractory B-cell malignancies.
Scale and accuracy of digital assays drives efficient DMTA cycles
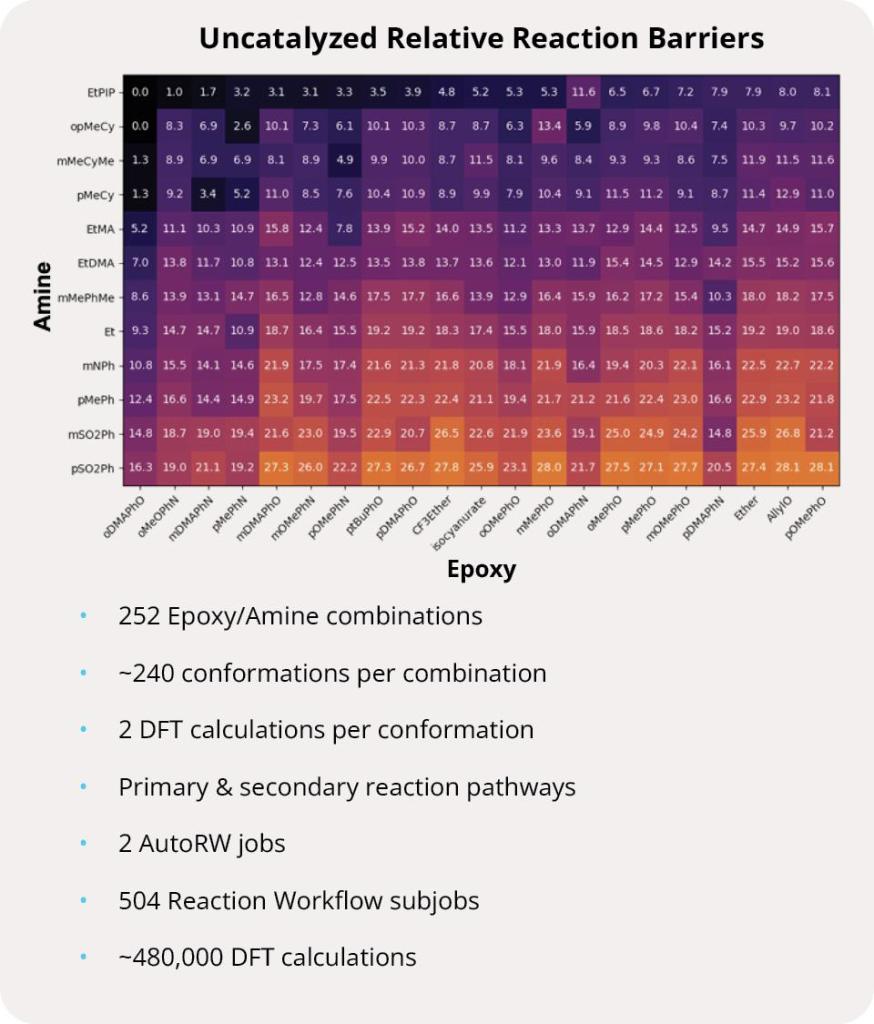
Finding a novel molecule with the right balance of on-target affinity and desired physicochemical properties is the essential challenge of every drug discovery program. In principle, increasing the number of rationally designed compounds assessed across these various properties increases the odds of success. Designing molecules in silico — with the speed and accuracy to traverse billions of molecules — is the guiding ethos of Schrödinger’s digital chemistry strategy. Specifically, this project combines rigorous physics-based modeling with machine learning (ML), predictive ADMET models, and data analytics to search and triage a chemical space consisting of more than 8B compounds. Ultimately, execution of this strategy enabled the identification of multiple novel series.
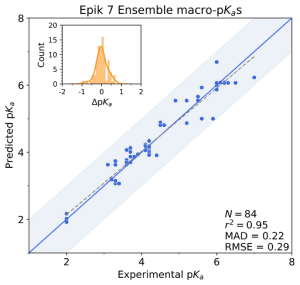
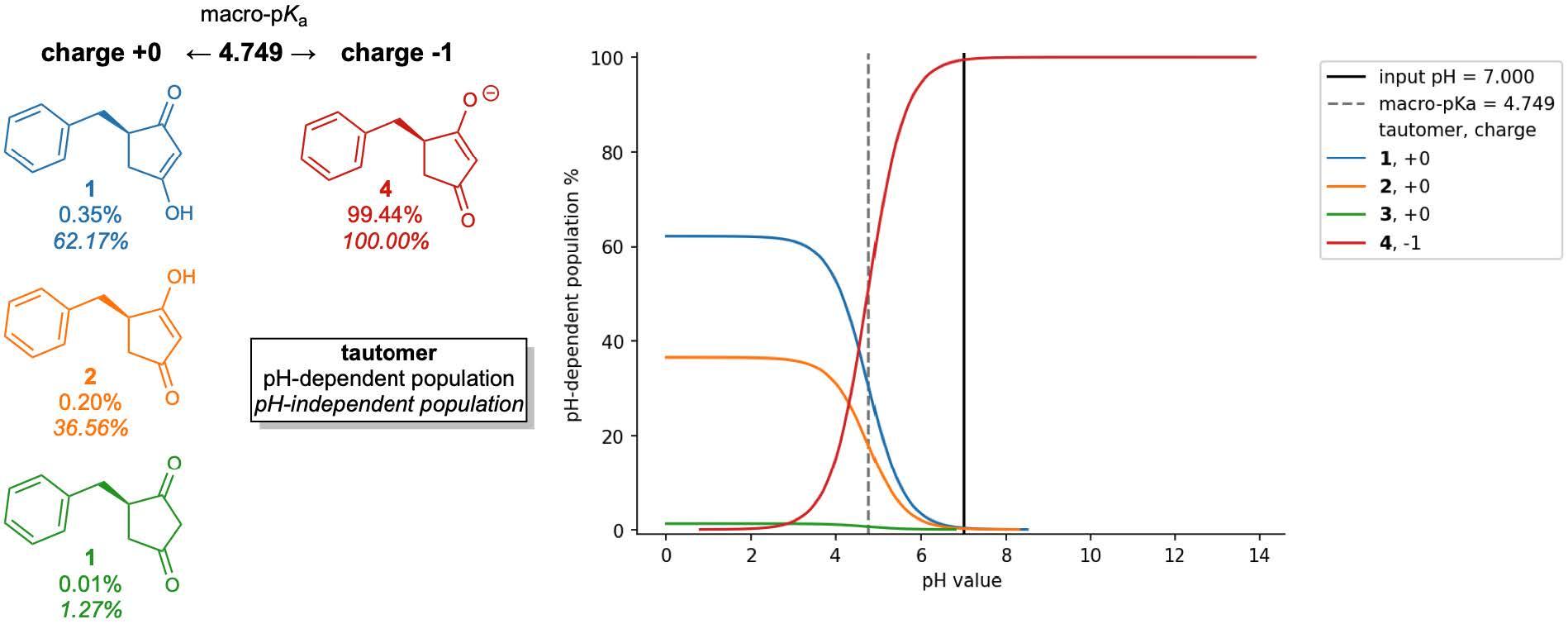
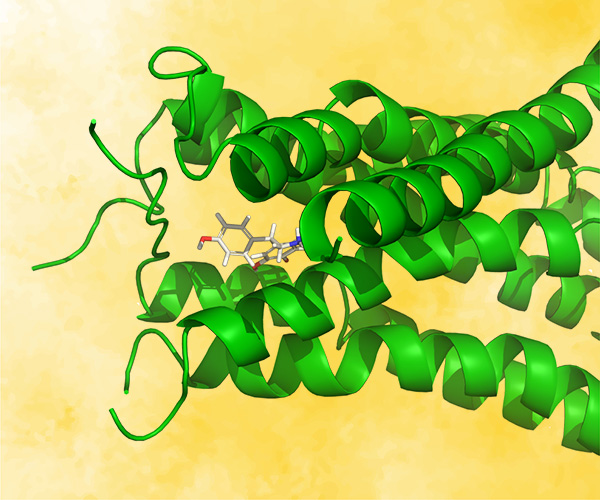
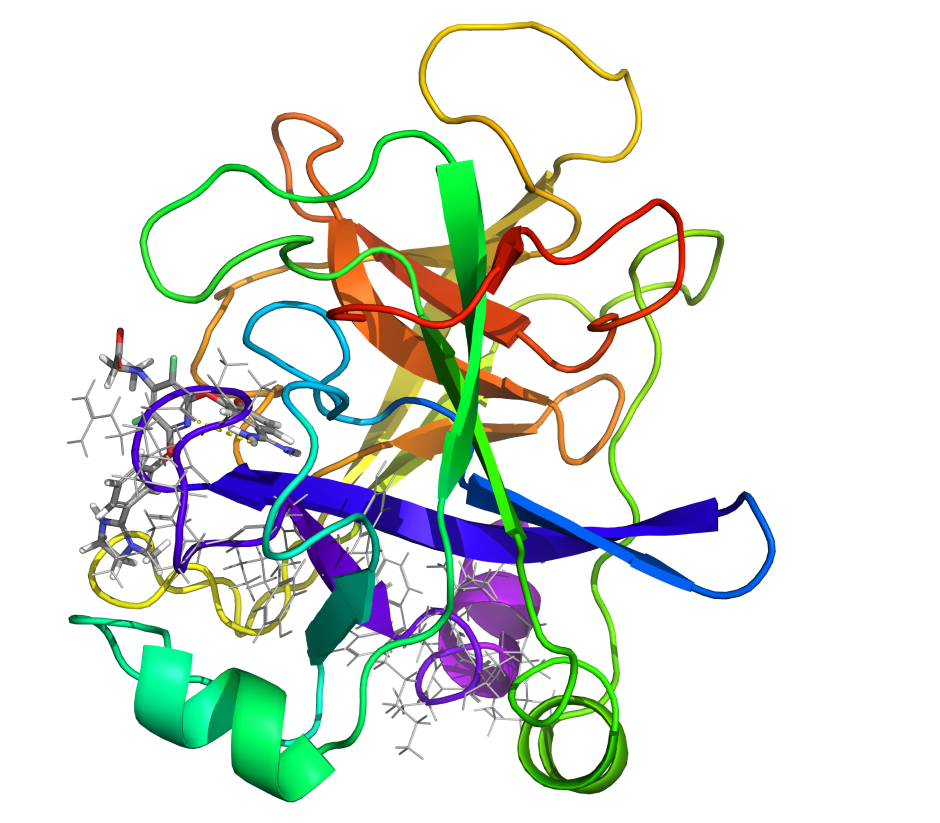
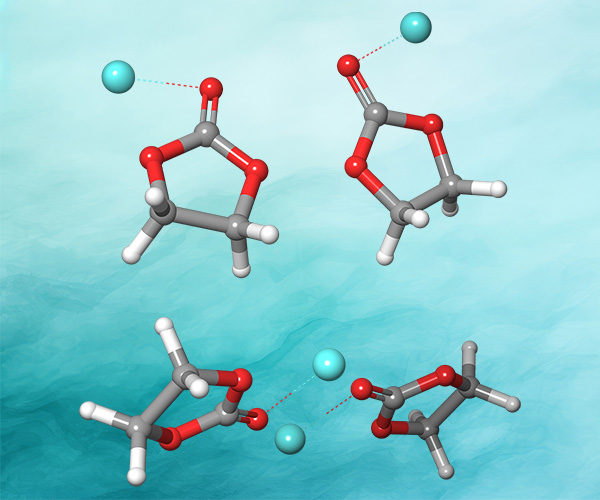
First, the team performed structure-activity relationship (SAR) analysis of existing chemical matter, followed by computational assessment of the allosteric binding site using WaterMap. As a result, the team identified a number of displaceable highenergy water molecules in regions of the binding site that provided an opportunity to gain potency while exploring different chemotypes.1 Schrödinger’s drug discovery team used this information to drive the evaluation of billions of compounds via a De Novo Design strategy for iterative large-scale design and scoring. This strategy included synthetically-aware, reaction-based enumeration, crowdsourced medicinal chemistry ideation, and FEP+ for free energy perturbation modeling. The accuracy and utility of FEP+ as a computational assay for the prediction of relative binding energies of molecules has been validated extensively, generating predictions within one kcal/mol of experimental values on average.2 By combining FEP+ with high performance cloud computing and machine learning (Active Learning FEP+), over 1,700 molecules were evaluated in the first three months of the project. All ideas and corresponding modeled data crowdsourced by the team were captured and analyzed with LiveDesign, a best-in-class, modeling-enabled collaborative enterprise platform for real-time project ideation (Figure 1). In less than three months, with fewer than 50 total compounds synthesized, the team was able to identify two novel and distinct series of highly potent MALT1 inhibitors, affording progression to in vivo testing.
Figure 1: Modeling strategy and design-predict-make-test-analyze (DPMTA) cycle employed for MALT1 inhibitor program, in which development candidate SGR-1505 was discovered in 10 months.
Overcoming the MPO challenge by tuning potency, solubility, and permeability simultaneously
Once potent chemical series were identified, the team focused on tuning physicochemical properties to meet the target product profile (TPP). They employed a multiparameter optimization (MPO) scoring system to triage molecules rapidly based on their predicted ability to satisfy the TPP. Calculation of the MPO score was based on values derived from predictive models for solubility, permeability, and potency. Using this strategy the design team assessed over 5,000 ideas and identified 43 compounds that met the program’s criteria. A handful progressed to synthesis and experimental testing, reducing cost and time significantly.
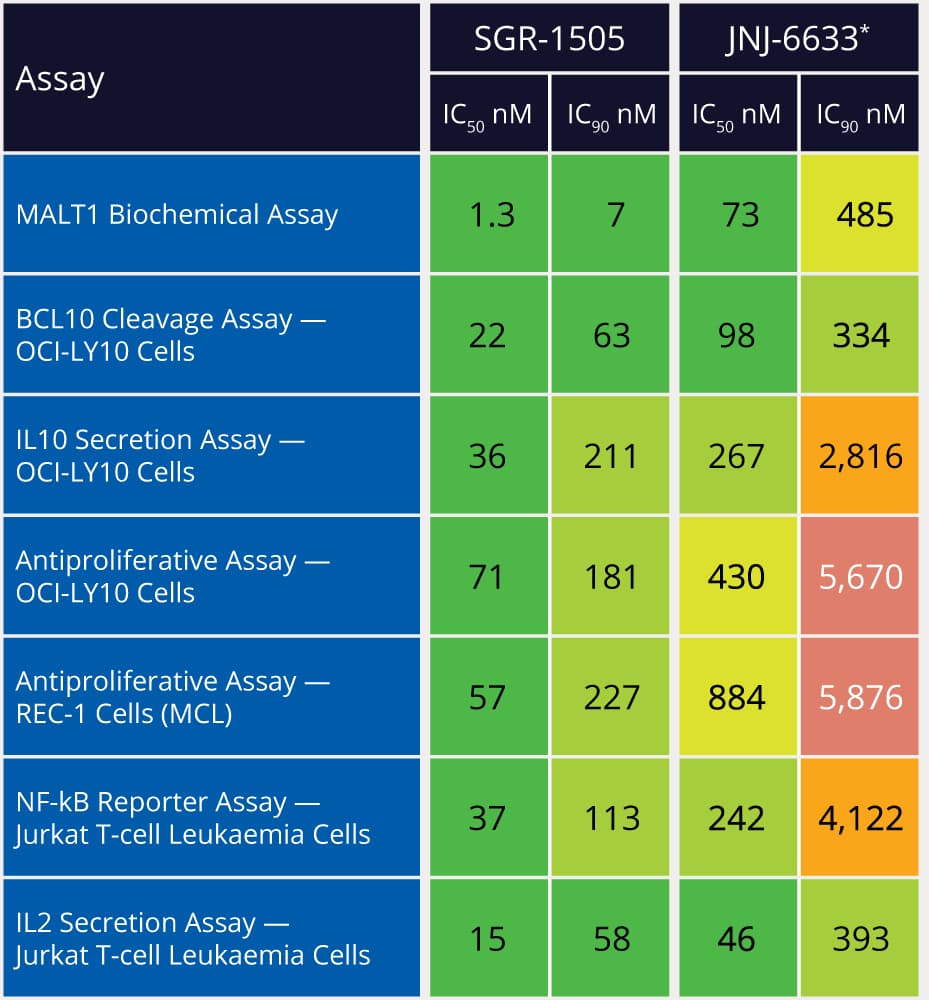
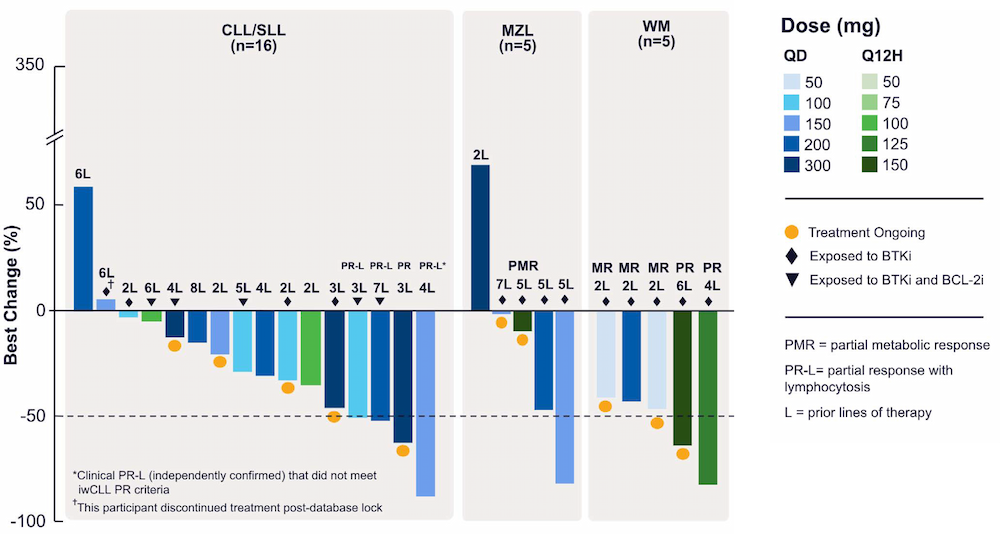
Within 10 months and a total of 78 compounds synthesized in the lead series (and 129 compounds program wide), the project team identified a potential best-in-class MALT1 inhibitor with balanced properties and on-target activity, SGR-1505 (Figure 2). In June 2025, SGR-1505 was observed in its ongoing Phase 1, open-label, dose-escalation study to have a favorable safety profile and was well tolerated, with encouraging preliminary efficacy in patients with relapsed/refractory B-cell malignancies.4 Responses were observed across a broad range of B-cell malignancies, including monotherapy responses in patients with chronic lymphocytic leukemia (CLL) and Waldenström macroglobulinemia (Figure 3).5
Enabling digital technologies to drive discovery programs
FEP+
Digital assay for predicting protein-ligand binding across broad chemical space at an accuracy matching experimental methods.
De Novo Design Workflow
Ultra-large scale chemical space exploration combining multiple compound enumeration strategies with an advanced filtering cascade.
WaterMap
Calculation of the positions and energies of water sites in a protein binding pocket.
LiveDesign
Collaborative enterprise informatics platform for centralizing access to virtual and wet lab project data and powerful computational predictions.
References
-
Calculating water thermodynamics in the binding site of proteins – Applications of WaterMap to drug discovery.
Cappel et al. Curr. Top. Med. Chem. 2017, 17(23), 2586-2598.
-
Advancing drug discovery through enhanced free energy calculations.
Abel et al. Acc. Chem. Res. 2017, 50(7), 1625–1632.
-
Characterization of potent paracaspase MALT1 inhibitors for hematological malignancies.
Yin et al. ASH Presentation 2021.
-
Schrödinger reports encouraging initial Phase 1 clinical data for SGR-1505 at EHA Annual Congress.
Schrödinger. 2025.
-
A Phase 1 study of SGR-1505, an oral, potent, MALT1 inhibitor for relapsed/refractory (R/R) B-cell malignancies, including chronic lymphocytic leukemia/small lymphocytic leukemia (CLL/SLL).
Spurgeon, et al. European Hematological Association Annual Congress. 2025.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Modeling Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.