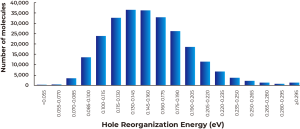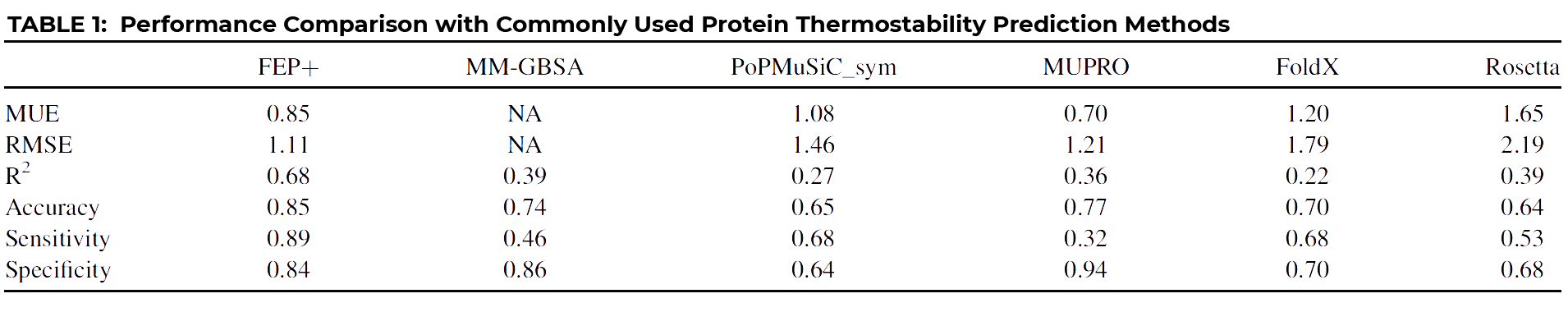Exploring the formulations of personal care products using a digital chemistry strategy
Share
Speaker
Jeffrey Sanders
Product Manager of Consumer Packaged Goods
Abstract
The demand for new and innovative personal care products is increasing due to changes in consumer trends and sustainability goals for CPG companies. Customers are becoming more discerning – choosing products based on understanding the ingredients and whether a product is made with natural or petroleum-based materials. These concerns highlight challenges for consumer goods research and development. To meet these challenges and retain their position in the consumer marketplace, new product formulations need to be developed and match the existing formulation’s key properties. Understanding how ingredients behave in products and “in action” will be necessary to drive not only new development but also end-to-end product tracing. To streamline this process, multi-scale physics simulations can be utilized to cut down product development timeline and cost, as well as optimize large-scale production by simulating digital twins. Molecular simulation provides a unique opportunity to predict how individual ingredients will behave in formulations. Atomistic simulations can help researchers and engineers understand product morphology, solubility, and other physical properties if the components are known. Unlike process simulations, only the chemistry and composition are required to build molecular models of up to millions of atoms and predict properties. Beyond physics-based modeling, chemical information can be used to build machine-learned models with existing experimental or sensory data. In this talk, we will show you molecular modeling in action and explore how digital chemistry strategies are driving innovation in personal care product formulations.
Learning Objectives:
How to gain insight of individual ingredient behavior and key properties of components in formulation using multi-scale physics simulations
How to predict key properties of formulations with advanced machine learning
How digital chemistry can accelerate your research and development in personal care product
A paradigm change in the design and optimization of OLED materials using a digital chemistry strategy
Share
Speaker
Hadi Abroshan
Senior Scientist
Abstract
Recent developments in device architecture of organic light emitting diodes (OLEDs) have opened a new avenue for innovative technologies to fabricate ultra-thin, flexible, foldable, and transparent displays. However, commercial advancement of OLEDs with higher performance requires continued discovery and development of novel optoelectronic materials. Given the enormous chemical design space available, traditional approaches based on chemical intuition and trial-and-error experimentation are expensive, time-consuming, and most often ineffective. A paradigm change in materials design and development is required to realize next-generation OLEDs.
In this webinar, we will present the impact of in silico technologies for systematic design, development, and selection of organic optoelectronic materials. Both physics- and machine learning-based approaches (e.g. active learning, goal-directed generative model) will be discussed. These computational approaches enable development of a better understanding of structure-function relationships from a molecular and morphological perspective. We also demonstrate accelerated OLED materials analysis through the combination of atomic-scale simulations with machine learning to pre-screen novel materials for high performance before laborious synthesis and device fabrication.
Key Topics Covered:
Understand the predictive capabilities of physics-based modeling in optoelectronics
Explore new machine learning capabilities for high-throughput screening to accelerate OLED materials discovery
Opening new worlds for structure-based drug discovery with advanced physics-based computational methods
Share
Speaker
Edward Miller
Director of Protein Structure Modeling
Abstract
The value of pursuing a structure-based drug discovery strategy has amplified in recent years as new highly predictive, physics-based methods have evolved and demonstrated the ability to accelerate the discovery of novel clinical compounds. However, these approaches are limited by the availability of high-quality structural models of the target protein.
Recent advances in structural biology such as cryo-EM and computationally predicted protein models (using machine learning and physics-based methods) have the potential to open a new world of targets to pursue. This webcast will describe how new advances in computational workflows are enabling structure-based drug discovery on these historically challenging targets and off-targets.
KEY TOPICS COVERED:
How new computational approaches can assist in building and validating high-quality protein structural models in the absence of experimental X-ray crystal structures
How these methods can be used to guide structure-based drug discovery programs, such as progressing hits from high-throughput screens, dialing out off-target liabilities, and improving on-target potency
What this means for the future of structure-based drug discovery amidst advances in structural biology
Schrödinger’s Materials Science software suite offers a range of computational solutions for advancing pharmaceutical formulation, from crystalline or amorphous form characterization, to selection of materials and excipients for processing, to formulations and delivery of active pharmaceutical ingredients (APIs).
Due to the accelerating pace of drug discovery, fast and efficient ways to both preformulate and formulate new drugs are critical elements of pharmaceutical development. The latest advancements in molecular modeling and AI/ML are enabling atomistic-level insights to improve drug formulations and the ability to evaluate large numbers of candidate materials and formulations prior to experiments.
Optimizing Drug Formulations with Machine Learning
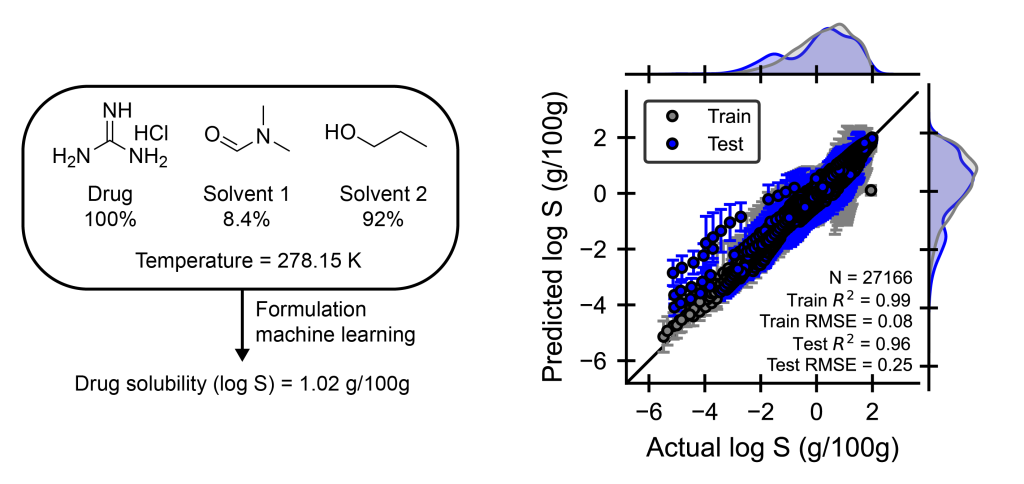
Mixtures of chemical ingredients, such as formulations, are ubiquitous in materials science, but optimizing their properties remains challenging due to the vast design space. Experimentally fine-tuning formulations for desired properties is expensive because of the large design space of both ingredient structures and compositions. Machine learning (ML) approaches that can accurately map ingredient structure and composition to properties offer a promising solution to rapidly screen formulations for desired target properties. Using Schrödinger’s automated Formulation ML workflow, we demonstrate that formulation-property models can accurately predict temperature-dependent drug solubilities for single or binary solvent systems. The parity plot shows that the Formulation ML workflow achieves a test set R2 of 0.96 (an ideal model would achieve R2 of 1.00), which highlights the accuracy of ML approaches. These tools enable rapid screening capabilities that transform the way we design drugs and take only seconds to generate a prediction, which is orders of magnitude faster than trial-and-error experimental exploration.1
Example of a drug in single or binary solvent mixtures with compositions in mole percent and temperature that is passed into a formulation machine learning model to predict the drug solubility in grams drug / 100 grams solution using a dataset extracted from Bao, Z, et. al. J Cheminform, 2024, 16, 117.
Accelerating Amorphous Solid Dispersion Development
Amorphous solid dispersions (ASDs) are widely used to formulate APIs into safe and effective media for human absorption. From screening for compatibility to understanding dissolution mechanisms, the Schrödinger Platform has tools for speeding ASD development.
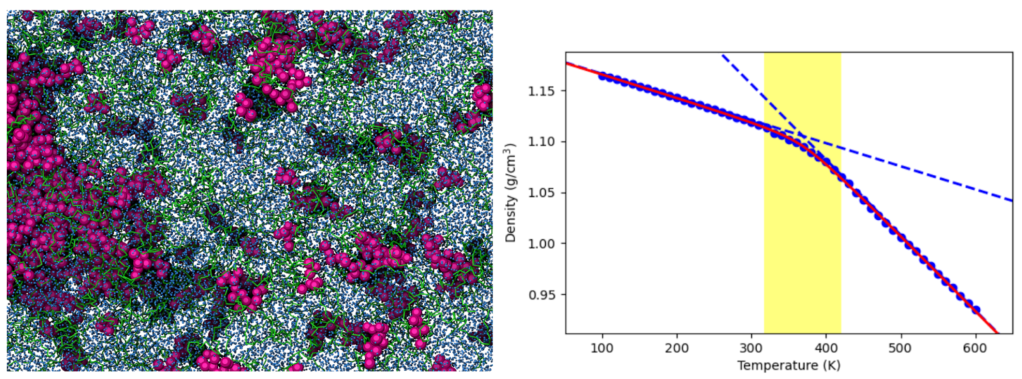
Complex interactions between an API and key ASD components (e.g., polymers, surfactants, and stabilizers) during dissolution are easily viewed and analyzed with coarse-grained physicsbased simulations such as dispersive particle dynamics (DPD) simulations. The underlying mechanisms governing the overall ASD dissolution process are then accessible, helping to solve practical challenges such as solvent-induced phase separation and the impact of drug load on ASD stability and miscibility. Beyond dissolution, other anhydrous dynamic processes are critical for ASD design. One key factor is the glass transition temperature (Tg), as maintaining the ASD system below Tg prevents excessive polymer mobility that could lead to API recrystallization, ultimately reducing ASD efficiency and shelf-life. Schrödinger’s molecular dynamics workflow provides a reliable method for estimating the Tg of the drug and ASD systems, allowing the targeted design of safe API ASD formulations.1-2
Molecular simulation applications for amorphous solid dispersions. Top: DPD simulations reveal polymersurrounding API aggregates in a ritonavir (purple balls) – copovidone (green tubes) ASD. Bottom: Schrödinger’s molecular dynamics workflow estimates the glass transition temperature (Tg) of bucindolol with a 3.6% error relative to the experimental value.
AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization
Share
Speaker
Karl Leswing
Machine Learning Tech Lead
Abstract
The lead optimization stage of a drug discovery program generally involves the design, synthesis, and assaying of hundreds to thousands of compounds. The design phase is usually carried out via traditional medicinal chemistry approaches and/or structure-based drug design (SBDD) when suitable structural information is available. Two of the major limitations of this approach are (1) difficulty in rapidly designing potent molecules that adhere to myriad project criteria, or the multiparameter optimization (MPO) problem, and (2) the relatively small number of molecules explored compared to the vast size of chemical space. To address these limitations we have developed AutoDesigner, a de novo design algorithm.
How digital molecular simulations will drive the next generation of innovation in reformulation and sustainability of consumer-packaged goods
Share
Abstract:
Molecular modeling has historically been viewed as a research tool with little connection back to commercial products. Computational power, expertise, and precise knowledge of chemical space, along with mismanaged expectations has limited the impact of molecular modeling in industrial settings until recently. With advances in physics-based simulation methods and machine learning, molecular simulation is quickly becoming routine alongside experimentation. In this talk, the utility of modeling to develop new products, rationalize product (mis)behavior, and understand how modeling can empower researchers to drive innovation will be highlighted. Case studies will be discussed that illustrate how modeling, when correctly applied, can provide novel insight into design and selection of surfactant-based formulations and interactions with packaging.
Jeffrey M. Sanders, Ph.D.
Product Manager and Scientific Lead of Consumer Goods
Jeffrey M. Sanders received his B.S. in applied physics from Worcester Polytechnic Institute and then his Ph.D. in biophysics and molecular pharmacology from Thomas Jefferson Medical College. Since joining Schrödinger in 2013, Jeff has served several roles in both the scientific and technical aspects of computational chemistry software. He is currently the technical lead and product manager for consumer goods.
With the advent of more powerful hardware and methods, the use of machine learning (ML) methods has seen a significant upsurge in chemistry-related applications recently. Specifically in drug discovery, the prediction of ADMET (absorption, distribution, metabolism, excretion and toxicity) properties is a main target for ML applications. Herein, we present performance metrics for Schrödingers automated ML model building engine, DeepAutoQSAR, on the ADMET subset of the Therapeutic Data Commons (TDC) — a large collection of public data for ML model building and benchmarking. We also compare the performance of DeepAutoQSAR to the performance of two open source projects, namely ChemProp and DeepPurpose.
DeepAutoQSAR is among the top-performing methods in 20 of the 22 investigated cases, clearly outperforming the other methods in 9 of those. For the other 11 cases, at least one of the other tested methods performs similarly. We believe that continuous development and further improvement of DeepAutoQSAR, in accuracy, robustness to chemical data shift and label efficiency will enable faster and more cost-effective means of drug discovery, ultimately leading to the introduction of novel therapeutics.
Introduction
It is widely recognized that the ADMET (absorption, distribution, metabolism, excretion and toxicity) profile of novel molecules plays a key role in the successful development of new drugs. This is reinforced by the amount of time and effort spent both in academia and the pharmaceutical industry to develop reliable models to measure and predict numerous related endpoints1. Due to the potentially catastrophic impact of an unfavorable ADMET profile in the later stages of drug development, a common goal is to identify potential issues as early as possible.
With the rise of ultra-large on-demand libraries and DNA encoded libraries (for example Enamine REAL Space or WuXi LabNetwork), early identification of liabilities requires methods that are computationally fast, cheap, and accurate enough to evaluate hundreds of millions of compounds without discarding potentially good candidates. This obviously precludes the use of experimental in vivo or even in vitro methods. Modern machine learning (ML) approaches, often coined artificial intelligence (AI), can easily process millions of molecules on short timescales and low computational costs with acceptable accuracy.
In contrast to physics-based in silico methods, ML/AI methods require high fidelity data to be trained to predict a given endpoint. High-quality training data is often unavailable; data need to be clean and well-curated, and datasets in chemistry applications are often smaller than those used in other domains like ML on images or text. These strict data requirements can limit the application of more complex ML/AI approaches since there is often insufficient amounts of training data to fit complex and accurate models.
However, recognizing the importance of profiling ADMET properties over the past decades, large pharmaceutical companies have generated a wealth of data which is often unfortunately non-public and exclusively applied for internal programs. Public data is rarer, but there are efforts to collect and aggregate public data 2 and also to share non-public data in smart ways to improve existing models while retaining data confidentiality 3.
The successes of deep learning (DL) approaches have led to a renaissance of ML/AI in chemistry applications, with a large number of both open-source and commercial software to pick from when targeting ADMET endpoints. While open-source software oftentimes can profit from faster development cycles and thus implements new scientific insights more quickly, application is often limited to domain experts. On the other hand, commercial software has the benefits of structured quality assurance (QA), documentation and support, and comes coupled with comprehensive user interfaces which significantly lower the barrier to entry for non-experts.
In this paper, we will take a closer look at the performance of two of the more popular open-source packages, ChemProp and DeepPurpose, and Schrödinger’s ML/AI package DeepAutoQSAR, demonstrating their comparative performance on a recently published set of benchmarks.
Hit to lead design of novel d-amino-acid oxidase inhibitors using a comprehensive digital chemistry strategy
Computational platform grounded in highly accurate predictive models enables team-based discovery of a novel chemical series engaging a complex CNS target.
Inhibition of D-amino-acid oxidase (DAO) has been hypothesized as a potential therapeutic strategy for schizophrenia. Schrödinger’s Drug Discovery Team engaged in a discovery effort with a collaborator to identify novel DAO inhibitors with potential best-in-class properties.
Program Challenges
Identify novel chemical matter while striving for best-in-class molecules that cross the blood-brain-barrier
Simultaneously optimize drug-like properties, improve CNS exposure, and affinity
Approach
The Drug Discovery Team deployed a large-scale digital chemistry strategy leveraging:
A centralized project data platform to facilitate knowledge-based medicinal chemistry design collaboration (LiveDesign, AutoQSAR)
Physics-based methods to predict affinity and prioritize design ideas for synthesis (FEP+)
Computationally-driven ideation and scoring workflow to amplify common enumeration strategies and screen hundreds of millions of compounds using machine learning coupled with physics-based free energy methods (FEP+, AutoDesigner)
Results
The team discovered a novel class of DAO inhibitors with desirable drug-like properties by confidently exploring synthetically-challenging chemistry. The team also identified a previously unexplored subpocket for further evaluation. The novelty of the compounds, coupled with well-balanced properties, demonstrates the extraordinary power of the approach to unleash project team creativity. By leveraging a digital platform, the team explored vast chemical space while simultaneously optimizing for drug-like properties in a challenging disease area.
Why use a digital chemistry approach?
A digital chemistry approach uses physics-based modelling, machine learning, and a team-based collective intelligence platform to design better molecules on accelerated timelines.
How to achieve optimal drug-like properties?
The development of CNS drugs poses several unique challenges. Fine-tuning physicochemical properties for optimal brain exposure is an essential element of CNS drug development. Many companies have discontinued neuroscience discovery because these challenges lead to longer development timelines and a lower probability of success.
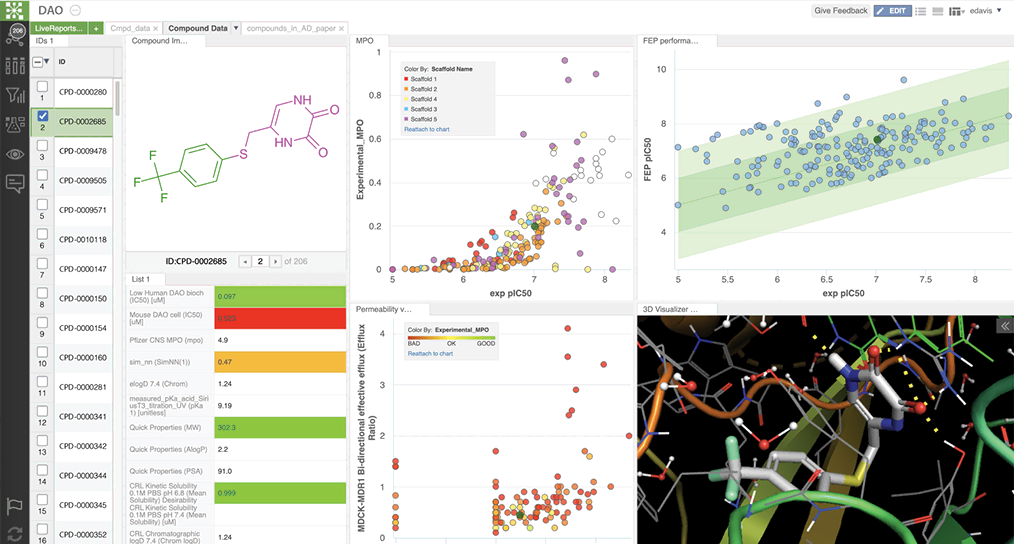
Schrödinger’s Drug Discovery Group developed property prediction models, using AutoQSAR deployed via LiveDesign, a web-based collaborative design platform. LiveDesign enabled teams of medicinal chemists to crowdsource designs and simultaneously optimize CNS properties with push-button workflows in a single interface (see figure 1). Compounds predicted in the desired property space were triaged using free energy perturbation (FEP+), a physics-based method for accurately predicting compound binding affinity. This workflow empowers teams to confidently pursue synthetically challenging compounds.
Figure 1. Schrödinger’s digital collaboration platform, LiveDesign, facilitates design optimization through custom multiple parameter optimization models by centralizing program data and improving team communication and collaboration.
How did a digital chemistry strategy enable improved hypothesis testing?
The team delivered high-quality molecules by working in an ecosystem that facilitated exploration of vast, novel chemical space and simultaneous optimization for desired properties through accurate physics-based modeling and machine learning.
The digital chemistry approach allowed the team to discover and quickly overcome many medicinal chemistry challenges in the pursuit of best-in-class molecules (see figure 1).2
Figure 2. SAR progression to achieve key milestones through late lead optimization with key compounds series represented. Crucial discovery and medicinal chemistry outcomes are highlighted. DHP represents dihydropyrazine and NHP, N-hydroxyl pyrimidine.
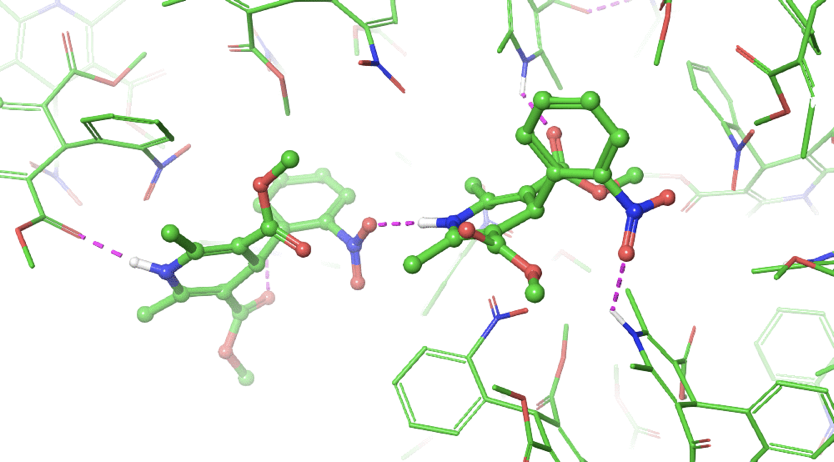
The team interrogated the atypical polarity of the DAO binding site, shown in panel A of figure 3. Chemists pursued challenging chemistry to reduce conformational flexibility and displace a high-energy water molecule by cyclizing and methylating cmpd 4 and 5 (see figure 2, panels B and C).
Finally, while literature and crystallographic structures suggested limited pocket volume for SAR exploration,3 FEP+ revealed the opportunity to interrogate this vector with larger chemical groups such as cmpd 6, as shown in panel D of figure 3.
Figure 3. A) Polarity of DAO binding site required a polar warhead. B) Cyclization of ligand linker reduced entropy improving affinity C) Displacement of high energy water near the binding site improved affinity (compare water present in panel B with panel C). D) FEP+ suggested exploring a novel subpocket predicted to improve potency (compare gray surface in panel A with the green surface in panel D).
How to interrogate vast chemical space and deliver novel chemical matter?
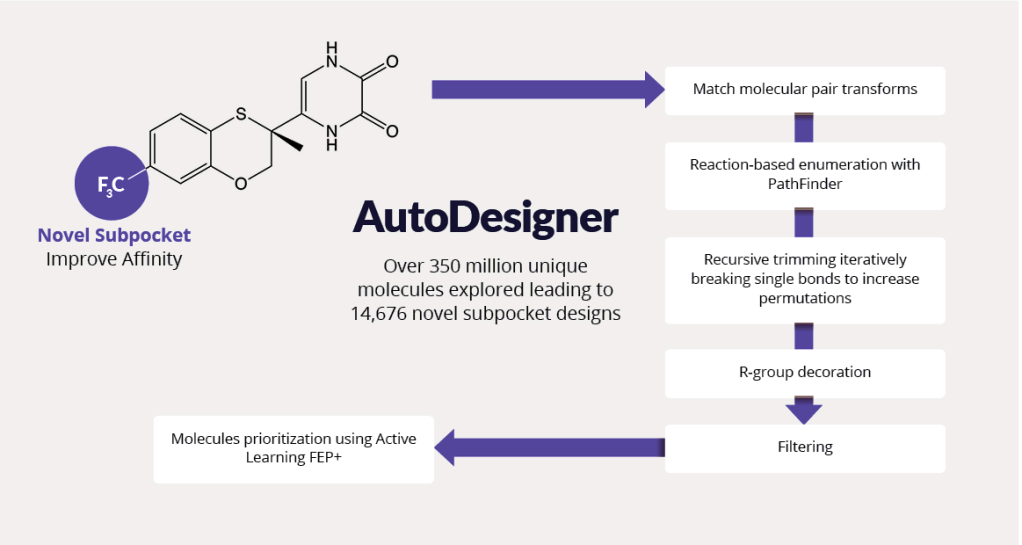
Pursuing best-in-class molecules required exploring vast chemical space outside of previously characterized drug-like molecules. The team utilized AutoDesigner, a multifaceted large-scale enumeration workflow (figure 4), to generate ideas exploring the novel DAO binding subpocket suggested by FEP+ (see figure 3, panel D).4
Figure 4. AutoDesigner enumeration and triage workflow explores SAR from the lead molecule while optimizing CNS drug-like properties to discover best-in-class molecules by covering vast chemical space.
To explore SAR and tune physicochemical properties, the team performed iterative cycles of AutoDesigner in the newly discovered subpocket. After triaging with appropriate filters, all molecular ideas were prioritized using free energy methods. The team trained active learning models using physics-based affinity predictions (FEP+) to prioritize compounds for synthesis. In total, more than 350 million ideas were generated and triaged.
What was the project impact?
Typically as drug discovery programs progress, teams struggle to balance desired properties, which leads to deficits in desired drug properties as novel scaffolds are explored and optimized.
Through a computational platform rooted in creative team collaboration, highly accurate predictive modeling, and enhanced by machine learning, a promising CNS DAO inhibitor series transitioned from hit discovery to lead optimization with approximately 11,000 compounds scored by FEP+ and only 208 synthesized. Of the 208 compounds synthesized, only 20 were inactive (>10μM) against DAO. By discovering novel compounds and concurrently performing multi-parameter optimization of critical CNS properties, the team delivered high project impact for this challenging disease area.
References
Bromet E.J., Fenning S.; Epidemiology and natural history of schizophrenia. Biol Psychiatry. 1999, 46 (7), 871–881.
Tang et al. Discovery of a Novel Class of D-amino Acid Oxidase (DAO) Inhibitors with the Schrödinger Computational Platform. ChemRxiv. Preprint. https://doi.org/10.33774/chemrxiv-2021-dkf1k.
Hondo et. al. 4-Hydroxypyridazin-3(2H)-one Derivatives as Novel d-Amino Acid Oxidase Inhibitors. J Med Chem. 2013, 56 (9): 3582-3592.
Bos et al. AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of D-Amino Acid Oxidase Inhibitors. ChemRxiv. Preprint.
Massive theoretical screening of organic semiconductor materials using cloud computing
Share
Panasonic scientists in collaboration with Schrödinger researchers published a paper in The Journal of Physical Chemistry, Massive Theoretical Screen of Hole Conducting Organic Materials in the Heteroacene Family by Using a Cloud-Computing Environment, about performing a large-scale theoretical screen of potential organic electronic materials in a cloud computing environment to determine molecules with optimal hole mobility properties. The performance of organic semiconductor devices depends crucially on the movement of charge through the weakly conducting materials composing the device. Positive charge carriers and negative charge carriers are holes and electrons, respectively. Charge transport can occur by hopping from molecule to molecule through the solid, modeled using atomic scale simulation techniques in the Schrödinger platform. The focus in this work was hole mobility in organic semiconductors.
Panasonic was interested in exploring structures with fused furans, thiophenes and selenophenes. These compounds are interesting for their use as organic semiconductors for a variety of applications. In particular, Panasonic is interested in making printable RF-ID (radio frequency identification) tags that require organic semiconducting materials with charge mobilities higher than currently available compounds. These materials could be used in housing, retail and warehouse management. Noncontact checkout and inventory would further enable future retail transaction technology. For example, a current Panasonic product, called ‘Reji-Robo’ , is a new automated checkout system that cuts labor cost by 10% while speeding up the process for customers.
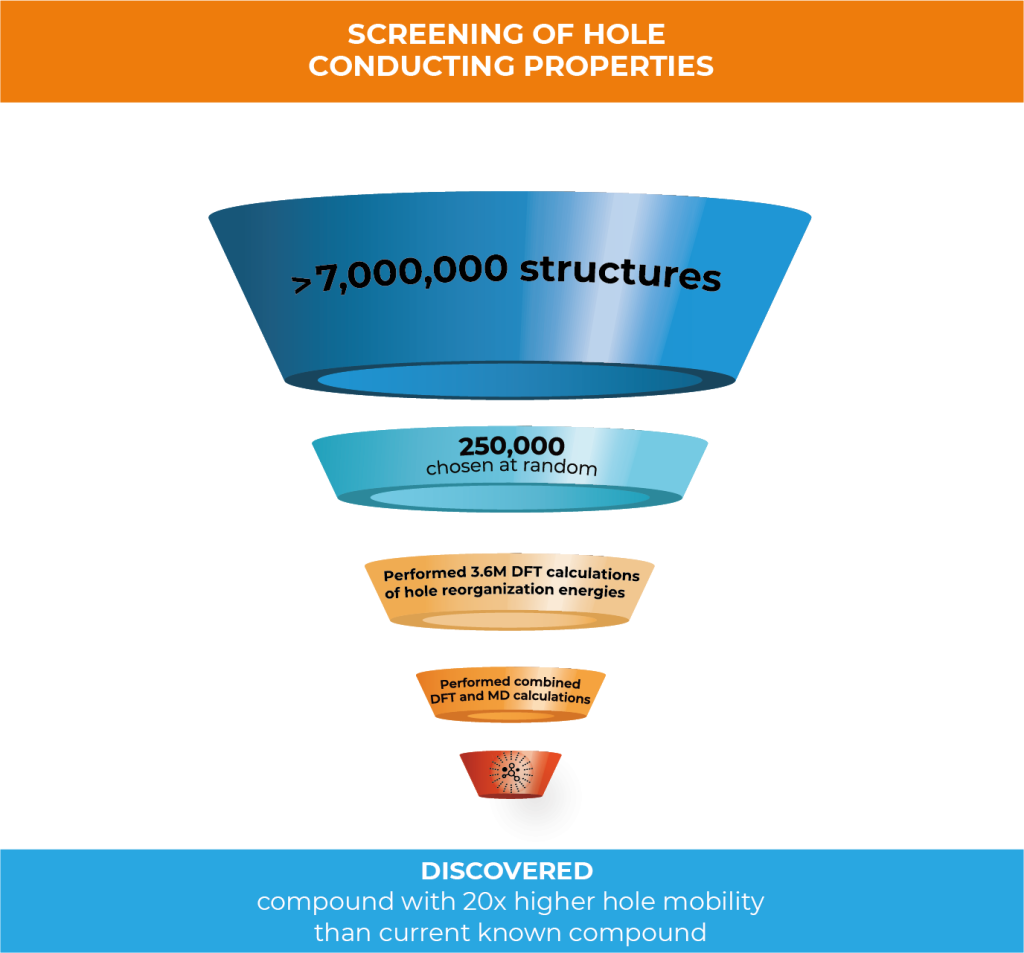
In this work, Schrödinger created 7,032,432 compounds through structural enumeration as the basis of chemical space for high-performance hole conducting materials. 250,000 of the initial set were randomly selected to perform density functional theory (DFT) calculations of hole reorganization energies. Because of the great amount of compounds to analyze, the team decided to perform the calculations in a cloud computing environment to drastically increase the throughput of calculations.
Leveraging Google cloud computing technology, 3.6M DFT calculations were run to predict hole reorganization energies of the 250,000 compounds—this was completed in just 16 days.
Alexander Goldberg, Senior Principal Scientist within Schrödinger’s Materials Science Group, explains the paper’s key findings:
Can you explain why this work is important?
Obtaining organic materials with improved mobility will help advance the field of organic electronics. Because organic electronic materials are a low-cost, highly flexible, easily processed, lightweight, and sustainable solution there has been a surge in both interest and investment in this field. The global organic semiconductor market alone is expected to grow to $179.4 Billion by 2024, up 22.4% from 2019.1
Although performance of organic material-based devices has improved to the point that it is now challenging inorganic materials for key applications, fundamental research is still needed for further development since the underlying physics is still unclear and working principles need to be clarified. We can utilize the Schrödinger platform to advance this research and enable design of high-quality, next-generation organic electronic materials.
Explain what this paper means. Why is it so exciting?
Performing a screening of this size in this amount of time wasn’t possible even just a few years ago. With the improvements seen in cloud computing, we’ve been able to increase the scale of using atomic scale simulation to screen chemical space drastically. When you consider that screening this number of compounds would take 100 days using a typical 200 CPU HPC resource, you can see just how much faster it is using cloud computing. If you were to screen this number using traditional experimental methods, it would take somewhere around 5,000 years.
In this project, you evaluated the suitability of 250,000 structures as hole transport materials from a chemical library of 7,032,432 structures to find structures with the most optimal hole mobility. Can you describe the process you used?
250,000 structures were randomly selected to perform density functional theory (DFT) calculations of hole reorganization energies. Then, the hole mobilities of compounds with the lowest 130 reorganization energy were further processed by applying combined DFT and molecular dynamics (MD) methods. By using this method, we were able to identify structures with a predicted hole mobility that is 20 times higher than dinaphthothienophene (DNTT)—a state of the art compound with one of the highest experimental mobilities observed.2
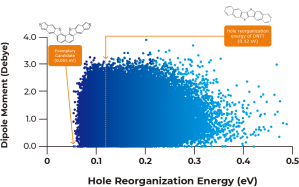
Figure 1: (a) Plot of the calculated dipole moments against calculated hole reorganization energies of the entire 250,000 compounds, and (b) histogram of the distribution of the hole reorganization energies through the entire library. Number of molecules with the reorganization energy being lower than 0.055 eV is 2, and that with the reorganization energy being 0.055 – 0.070 eV is 227.
As described in Figure 1A above, calculated dipole moments are plotted against hole reorganization energies for the 250,000 compounds. Good charge transport materials should have small dipole moments to avoid charge trapping in the solid state. The hole reorganization energies range from 0.055 to 0.50 eV. From there, we narrowed down to 17,000 compounds that had hole reorganization energy values below 0.1 eV, as shown in Figure 1b. When you compare that to DNTT which has a hole reorganization energy of 0.12 eV, these 17,000 compounds could all be judged as promising candidates for improved hole transport materials.
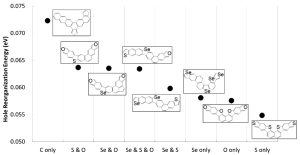
Figure 2 shows the minimum calculated hole reorganization energy in every considered topological compound together with their corresponding chemical structure.
Figure 2: (Figure 6 in publication): Minimum calculated hole reorganization energy for each category of compounds together with their corresponding chemical structure.
Was there a stepwise selection carried out to filter for high mobility candidates?
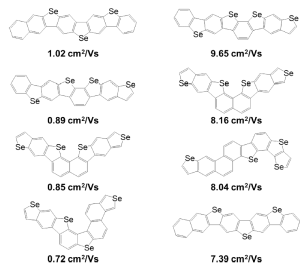
Yes. For example, because the cost of calculating mobility is much higher than calculating reorganization energy, after the massively parallel DFT calculations on the cloud, we selected 130 compounds with the lowest hole reorganization energy for further calculations to obtain estimated condensed phase hole mobility. By calculating 130 compounds we were able to rank which ones had the highest hole mobilities. Figure 3 (below) shows the chemical structures of compounds with the top four highest hole mobilities from that group. Two methods based on different approximations were used to compute mobility in ranking the candidates. Percolation theory is based on a random walk approach, whereas energy disorder theory is based on the distribution of energy states in the solid.
Figure 3: (Figure 10 in publication). Chemical structures of compounds with the top four highest hole mobilities using two different methods for calculating mobility, percolation theory (left) and energy disorder theory (right).
Are there other examples of how Schrödinger uses cloud computing?
Schrödinger utilizes cloud computing not only in Materials Design, but also in our Drug Discovery and Biologics research. Cloud computing enables us to screen vast areas of chemical space in a fraction of the time it would take with on-premise computing resources.
Schrödinger has a history of breaking records for virtual screening in the cloud. In 2013, our materials science team partnered with Cycle Computing, creating a 156,000+ core cloud computing run that was dubbed the Megarun.3 It used virtual machines across eight regions of Amazon Web Servcie’s public cloud around the world for a total of 18 hours and a total cost of just $33,000. It was the largest and fastest cloud computing run at that time in history. When you look at that milestone compared to this screening, you can see how quickly cloud computing technology is advancing.
Earlier this year, Schrödinger partnered with Google Cloud to accelerate our research. With nearly limitless processing power on demand, we are able to explore a larger area of chemical space and model vastly more compounds in a fraction of the time it would take using on-premise computing.
Google also donated resources to advance our work in the fight against COVID-19. Schrödinger is part of a philanthropic initiative with leading biopharma companies from around the world to develop antiviral therapeutics. Google Cloud is providing 16 million hours of GPU time as part of this collaboration. It’s equivalent to accessing the world’s fastest supercomputers.
What’s next?
Our next step is to continue our close collaboration with Panasonic and use the results from this study to enable machine learning techniques to determine structure-property relationships and further screen organic semiconductor design space for high mobility application.
Any final thoughts?
Cloud computing has revolutionized how we do research. Atomic scale simulation is already making a tremendous impact on materials R&D, lowering cost, shortening timelines, reducing risk and driving innovation. Cloud provides essentially, “limitless” resources. We no longer have to limit our pool of candidates to specific libraries. Being able to access the expanse of chemical space means we have a better chance of finding the best candidates with the most optimal properties. And, being able to do it faster means we are able to get results quicker that inform what next steps are needed. It’s an exciting time to be working in materials research.
About the Author
Alexander Goldberg
Ph.D., earned his Ph.D. in Chemical Physics from Tel Aviv University in Israel
Dr. Goldberg has been with Schrödinger for 8 years. His personal research focused on absorption, emission and IR spectroscopy of small clusters. He was also involved in the development of photochemical reactions of organic molecules to be employed in optoelectronic devices as well as finding materials for hydrogen storage in fuel cells and Li battery technology. His research was based on molecular simulations at different length- and time-scales using computational methods of quantum mechanics, classical physics and mesoscale modeling.
References
Organic Semiconductor Market Report
Market Research Future; Published September 2020
$68 million, 200-year, 150,000-core analytics job run on Amazon’s cloud in 18 hours for $33,000
Refining the route to solubility: Publication showcases a new FEP+ method to predict solubility prospectively in live drug discovery projects
In collaboration with Janssen and Nimbus, Schrödinger published a preprint in ChemRxiv, A Free Energy Perturbation Approach to Estimate the Intrinsic Solubilities of Drug-like Small Molecules. The paper presents a novel physics-based approach to predicting the aqueous solubility of small molecules that takes into account three-dimensional solid-state characteristics in addition to polarity. The method performed well not only on public datasets, but in testing against the measured solubilities from a large number of advanced programs in-house at Janssen. Furthermore, the paper reports on the first prospective application of the method in a live drug discovery project in collaboration with Nimbus Therapeutics.
Bringing together FEP+ Solubility, FEP+ potency, RRCK passive permeability modeling, and physico-chemical parameters like PSA in a modeling screening funnel can substantially improve the probability of success and accelerate the project timeline.
Dr. Mondal explains the paper’s key findings:
Tell us what’s the key advance in this paper, and why is it important?
In this paper, we introduce a brand new technology, FEP+ Solubility—a physics-based method that provides a detailed examination of the 3D solid-state packing structure of a molecule and accurately predicts solubility without a training set. FEP+ Solubility is built on our free energy perturbation method, FEP+, and is both accurate and has a wide domain of applicability, making it particularly suitable for drug discovery research, where exploring novel chemical space could be crucial to success.
Aqueous solubility is a focal point during lead optimization in drug discovery because it affects bioavailability, a molecule’s ability to enter the bloodstream and circulate throughout the body. Given that it is estimated that almost 40% of all new chemical entities (NCE) are determined to be insoluble, or nearly so, and that molecules with low solubility often experience issues during clinical and pre-clinical trials, leading to failure, it becomes clear why improving solubility is often a focal point during lead optimization. Moreover, straightforward attempts to increase solubility during lead optimization can degrade membrane permeability and/or potency.
How did researchers traditionally try to improve solubility? And how does FEP+ Solubility compare with previous approaches?
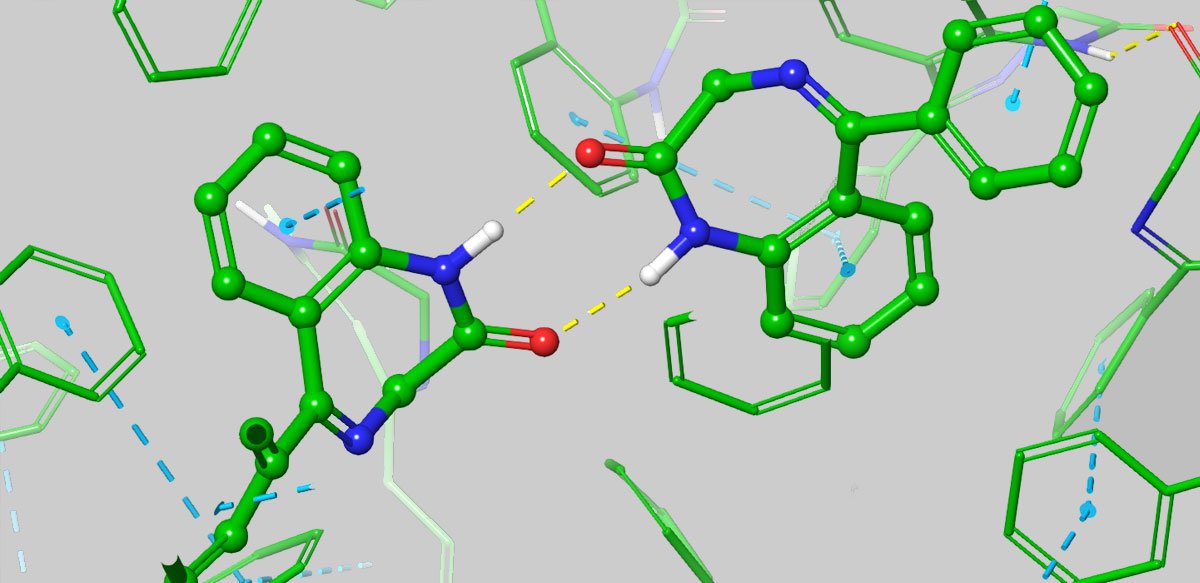
The most common approach to improving solubility was empirical—guided usually by logP or sometimes by machine learning models fitting existing data of experimentally measured solubility to various molecular descriptors, such as logP and polar surface area. However, such empirical approach can actually lead one astray, for example, simply adding polar groups can preferentially stabilize the solid state, and lead to the sometimes counterintuitive result of reducing solubility. FEP+ Solubility, on the other hand, allows for a detailed computation of the 3D solid-state packing energetics, providing insightful predictions about interactions that can improve or degrade solubility. A great example of this is Benzodiazepine, where it has been shown that substitutions that disturb favorable solid-state interactions can lead to improved solubility.
From Figure 3B in paper: Stabilization of the aggregate via H-bonding involving a polar substituent can nullify any intended solubility enhancement from the polar piece, as seen in the 3D structures of the aggregate from a simulation snapshot of benzodiazepine, where H-bonding between the uncapped NH of the cyclic amide with the carbonyl from an adjacent molecules add to the stabilization of the aggregate from intermolecular pi-stacking.
Another key difference between empirical methods and FEP+ Solubility is that the former requires a training set, and thus is inherently limited in its applicability to the chemical space of the training set, and often struggles to extrapolate to novel chemical entities. Unfortunately, it’s often necessary to expand beyond this limited scope of chemical space in order to achieve the desired solubility in balance with other parameters or IP, and this is where FEP+ Solubility really shines with its large domain of applicability. Furthermore, the understanding gained from FEP+ Solubility studies such as elucidating an in silico solubility SAR, can help the project team ideate novel chemical structures to improve solubility.
Can you speak to the performance of this new method, FEP+ Solubility, and how you are using the technology?
Sure, we did extensive retrospective studies to test our model against available published solubility data for compounds of pharmaceutical interest, and saw great agreement. But more importantly, we began to use the model in prospective studies, and again we saw excellent performance by FEP+ Solubility in classifying compounds based on their solubility profile, making it a quotidian tool in our arsenal.
As part of Schrödinger’s Drug Discovery Group, we use the Schrödinger platform in all of our internal and collaborative drug discovery projects. The platform consists of three things: advanced physics-based methods, machine learning, and enterprise informatics.
The predictions from various physics-based methods including FEP+ Solubility and machine learning methods, where applicable, are captured within LiveDesign, our web-based enterprise informatics program, facilitating synthesis decisions based on a holistic comparison of a large number of ideated molecules. The most promising compounds emerging from each round are then further optimized through additional cycles of computational analysis enabling us to determine the best candidates for synthesis.
FEP+ Solubility sounds like a significant advancement, how do you see it impact drug discovery projects?
Drug discovery teams often find themselves trapped in lengthy and uncertain cycles of optimization where they change R-groups to improve solubility, only to lose ground against potency, or an ADME property like permeability; or vice versa. These parameters need to be satisfied in the same molecule but are generally anti-correlated, meaning that if we are limited to designing along the dimension of polarity / hydrophobicity alone, often it would be to the detriment of other important properties.
By going beyond just polarity, it becomes easier to overcome the potency / selectivity / permeability / solubility tradeoffs to obtain higher quality chemical matter faster. With FEP+ Solubility used in conjunction with FEP+ analyses of potency and selectivity, and RRCK passive permeability modeling at scale, it becomes possible to break out of insoluble regimes and achieve balanced profiles within 1-2 cycles of synthesis.
This is especially true for projects where most synthesized compounds are found to be insoluble, the probability of success for meeting the Development Candidate guidelines can be dramatically improved by focusing synthesis on high quality chemical matter predicted to be soluble, potent, selective, and permeable.
The value of using FEP+ for solubility is particularly high when using it together with FEP+ for potency and selectivity, and RRCK permeability modeling, because the net outcome is more than the sum of the individual analysis. For example, one may decide to remove an aromatic ring based on FEP solubility results; however, if the aromatic ring is productive in the binding site, the probability of success is much higher if FEP+ potency modeling is used to maintain the potency while replacing the aromatic ring.
Our modeling platform allows researchers to map out the SAR for potency, selectivity, permeability, and solubility side-by-side for different series and sub-series. This information can help teams strategize which core to prioritize for further optimization, how to functionalize a given core, and to stimulate the iterative design of new ideas.
Any final thoughts?
To summarize, our approach through the FEP+ framework provides an opportunity to systematically predict the solubility of novel molecular entities. Beyond early stage drug discovery, this work lays down the foundation for solving key problems in formulation and materials science contexts with computational modeling. That is a world of impactful advances waiting to be made!
Tell us what was the main motivation behind this study?
The function of a protein is tightly coupled with its structure and dynamic behavior. Therefore, understanding the thermostability of proteins can provide fundamental insights into how they work. For example, single point missense mutations may directly affect protein function, leading to diseases. Many of these mutations have been found to be linked to protein thermostability. Also, from a practical perspective, protein stability engineering has wide applications in various industries, for example, vaccines and antibodies need to be engineered to have long shelf life and prevent aggregation; industrial enzymes for food, detergents, paper, or fuel need to be designed to be stable and functional in the desired environments.
As you know, we’ve had tremendous success in using our FEP+ technology in advancing small molecule drug discovery—we’ve published many papers, and all of our drug discovery projects have achieved milestones at an accelerated pace when compared to traditional approaches. But what’s less well established is how well FEP+ could advance projects involving biologics, so we set out to design a study to specifically examine the performance of FEP+ in predicting protein thermostability. We aimed to assess both quantitatively and qualitatively the technology as a viable option to predict changes in protein thermostability upon mutations such that it may guide protein engineering projects.
Had there been previous studies using FEP on proteins? And if so, how does the current study differ?
The idea of applying free energy perturbation theory (FEP) to predict the effect of single point mutations on protein stability is not new; however, production-level calculations to predict protein stability in industrial settings have not been possible due to a couple of severe limitations, including the inability to model mutations to and from prolines, where the bonded topology of the backbone is modified, and the complexity in modeling charge-changing mutations. In this study, we extended the FEP+ protocol to accurately model both of these types of mutations. Furthermore, because FEP calculations could be computationally intensive, many earlier studies were limited to very short simulations, and with limited solvation around the mutation, ultimately resulting in less accurate predictions. Thanks to algorithmic improvements and hardware advances in GPUs, Schrödinger’s FEP+ has demonstrated repeatedly that it can meaningfully impact projects within realistic timelines.
Because we set out to specifically assess FEP+’s performance in dealing with the challenging proline and charge-changing mutations, we paid particular care to selecting the test systems so that we could quantitatively evaluate our results – we curated a data set with high-resolution crystal structures for both wild-type and mutants, and with accurately measured stability data. You may find details of our test systems in the paper. And I’m happy to report that we achieved comparable accuracy in predicted stability changes as observed previously in countless small molecules studies, demonstrating that FEP+ is indeed an appropriate computational strategy for studying biologics.
Are there other methods for predicting protein thermostability? And if so, how do the present FEP+ results compare?
Yes, because of the importance of being able to accurately predict protein stability, many different methods have been proposed over the years. We compared our FEP+ results with several of the more popular methods.
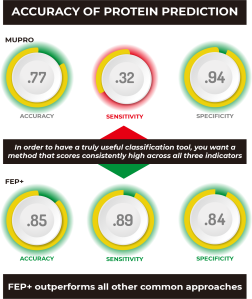
As you can see at a glance, FEP+ outperforms all other common approaches quantitatively, with the possible exception of MUPRO, which is a machine learning model using only sequence information and all mutations as part of its training set. However, it’s been suggested that MUPRO may suffer from training set bias, and indeed this is born out when you examine the qualitative measures of Accuracy, Sensitivity, and Specificity, also given in the table.
Let me explain what these three measures mean by using an example:
if experimentally it’s found that 90 out of 100 mutations are destabilizing, and one blindly guessed that all mutations are destabilizing,
then one’s guess would score 90% for Accuracy,
but would score 0% for Sensitivity, which measures the ability to correctly identify stabilizing mutations,
and 100% for Specificity, which measures the ability to correctly identify destabilizing mutations.
In order to have a truly useful classification tool, you’d want a method that scores consistently high across all three indicators, like FEP+, which clearly outperforms all other methods, including MUPRO, which while scoring well for Specificity, owing to its training set, it nevertheless is biased by that training set to be a poor predictor of stabilizing mutations, as indicated by its very low Sensitivity score of 0.32.
But isn’t FEP+ computationally expensive? How well can it be deployed at scale to meaningfully accelerate a discovery program?
It is true that FEP+ studies require a computational investment, but thanks to software algorithmic improvements and the ever-increasing processing power of GPU hardware, full analyses using FEP+ is well within reach for real-life projects. Furthermore, as described in our paper, we devised a screening cascade taking advantage of the much more computationally efficient residue scanning, available within our BioLuminate application, which tends to err on the side of false positives, as a filter for further FEP+ analyses. This way, one could rapidly screen all relevant mutations, and only pass onto FEP+ those that are found to be stabilizing, and let FEP+ identify those mutations that are most likely to be true positives.
That all sounds very promising, are there future plans for enhancing or expanding the technology?
Yes, of course, we’ve learned a great deal from this study, such as how to best model the protein’s unfolded state to achieve higher accuracy in the predicted change in stability. We also scrutinized our results and identified potential cases that may require longer simulations to fully capture large structural changes. We will continue to refine the protocol to improve the performance both in terms of prediction throughput and accuracy.
Author
Jianxin Duan
Director, Applications Science, Schrödinger
Jianxin Duan, Ph.D., is a Director with Schrödinger’s Applications Science group. He earned his Ph.D. in Biophysics from Karolinska Institute in Stockholm. Dr. Duan has been with Schrödinger for 14 years. He was involved in the development of ligand based methods, including cheminformatics, pharmacophore modelling and machine learning. His current personal research focuses on protein and antibody design.
Chinese webinar: Polymer innovation with computational chemistry
Share
Speaker:
Dr. Yuling An, Product Manager for Materials Science Machine Learning and Enterprise Informatics
Abstract:
Polymers have found use in industries from aerospace composites to food packaging to drug delivery. Their usefulness is tied to the intersection of chemistry, macromolecular structure, and microscale behavior. As the use of polymers has grown so has the need for better tools to predict and understand how the chemistry, processing, and macromolecular structure impacts performance. This webinar will review computational chemistry developed for polymers and how it has become a practical tool for polymer research engineers and scientists to have in their toolbox.
Polymer property predictions using chemically informed simulations
Computational chemistry techniques for thermosets and thermoplastics
Examples in area of epoxy resins, polyacrylates, polyolefins
Techniques for integrating simulation into industrial research and development