
Develop cleaner, safer energy materials with digital chemistry
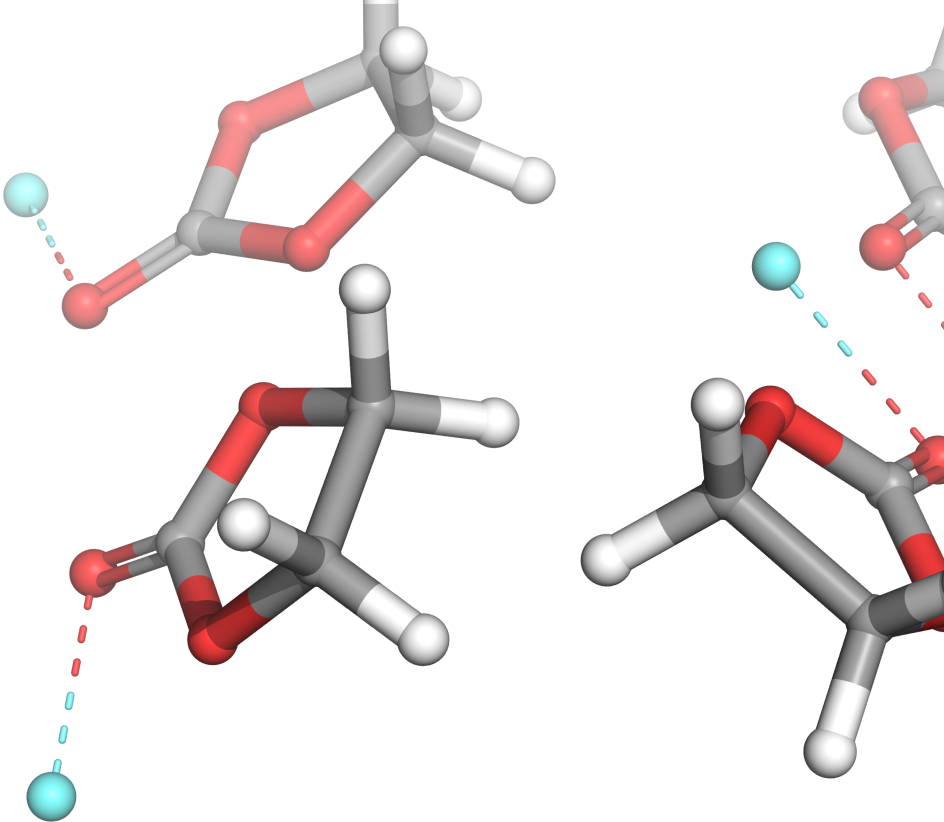
Discover and optimize energy materials at the molecular level
Safer, cheaper, and more effective batteries, fuel cells, and supercapacitors are critical in overcoming societal ecological challenges in the automotive, aviation, and energy industries.
Schrödinger’s Materials Science platform provides the tools to model materials at the molecular level, using computational power to drive forward the development of cleaner, lighter, safer, more energy-efficient, and lower cost materials for batteries, fuel cells, and photovoltaics – ready to power the next generation of innovation.

Intuitive computational workflows designed by energy materials experts

Easy-to-use system builders for all material types
Powerful workflows for physics-based simulation, machine learning, and data analysis
Dedicated customer support and extensive training resources
Your toolkit for energy materials innovation
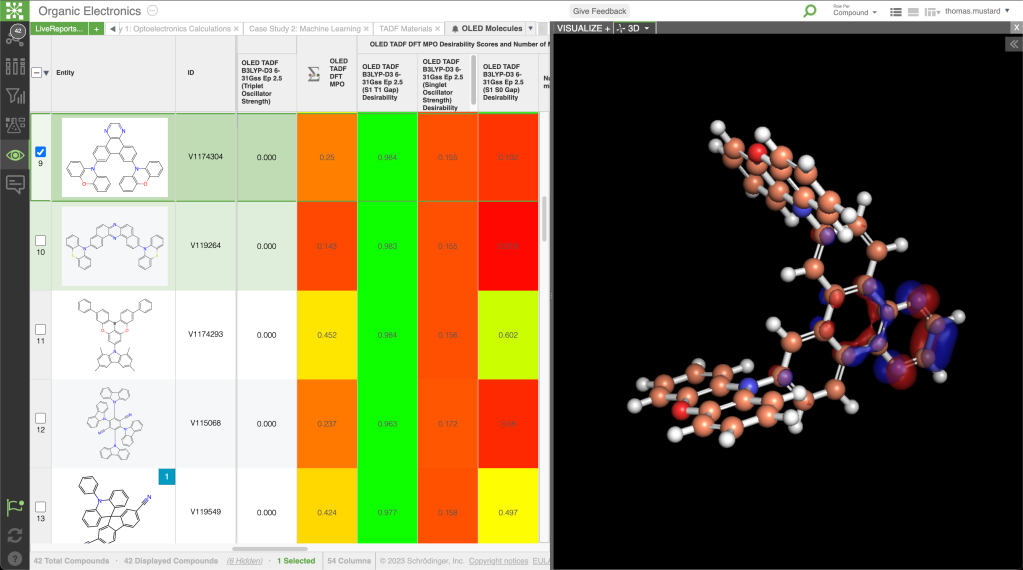
Predict key properties for batteries, fuel cells, photovoltaics, and hydrogen storage R&D
- Explore electrode, electrolyte, and solid electrolyte interphase (SEI) properties such as redox potentials and ion mobility (diffusivity and coordination environments) for battery materials
- Optimize photovoltaic material properties and performance metrics for semiconductors, photosensitive materials, perovskites, and organic photovoltaics
- Elucidate chemical reaction profiles for energy storage processes, catalytic mechanisms, and degradation pathways
- Predict hydrogen (or other small molecule) molecular mobility and stability in storage materials
Accelerate new materials discovery with high-throughput screening and machine learning
- Run high-throughput screening of new materials candidates to identify the best performers
- Assess new catalysts for energy-related transformations, such as electrolytic hydrogen production
- Screen electrolyte properties relevant to SEI formation
Enable access to digital materials design through a centralized informatics platform
- Bridge the gap between experimental and computational data
- Drive faster and better materials design with real-time access to predictive models
- Enhance collaboration and decision-making across your enterprise


Case studies & webinars
Discover how Schrödinger technology is being used to solve real-world research challenges.
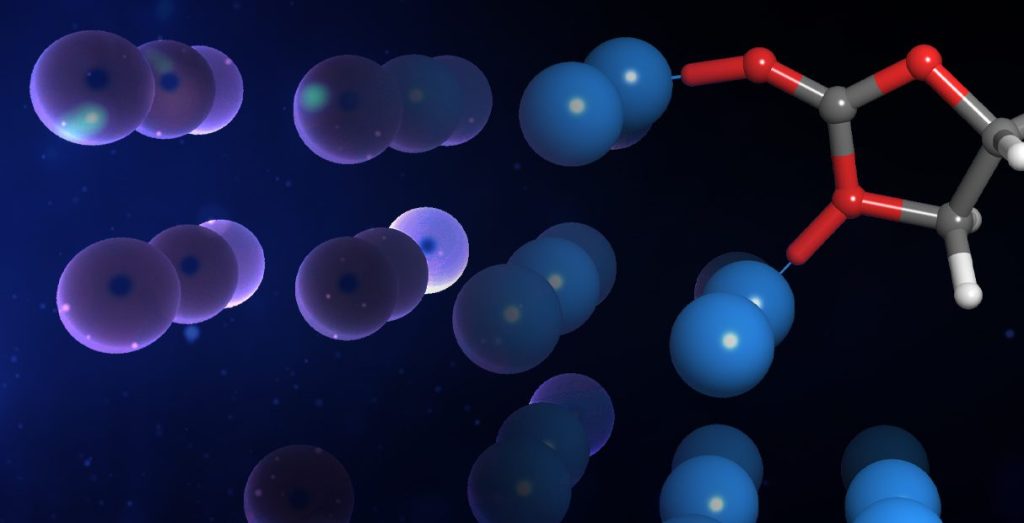
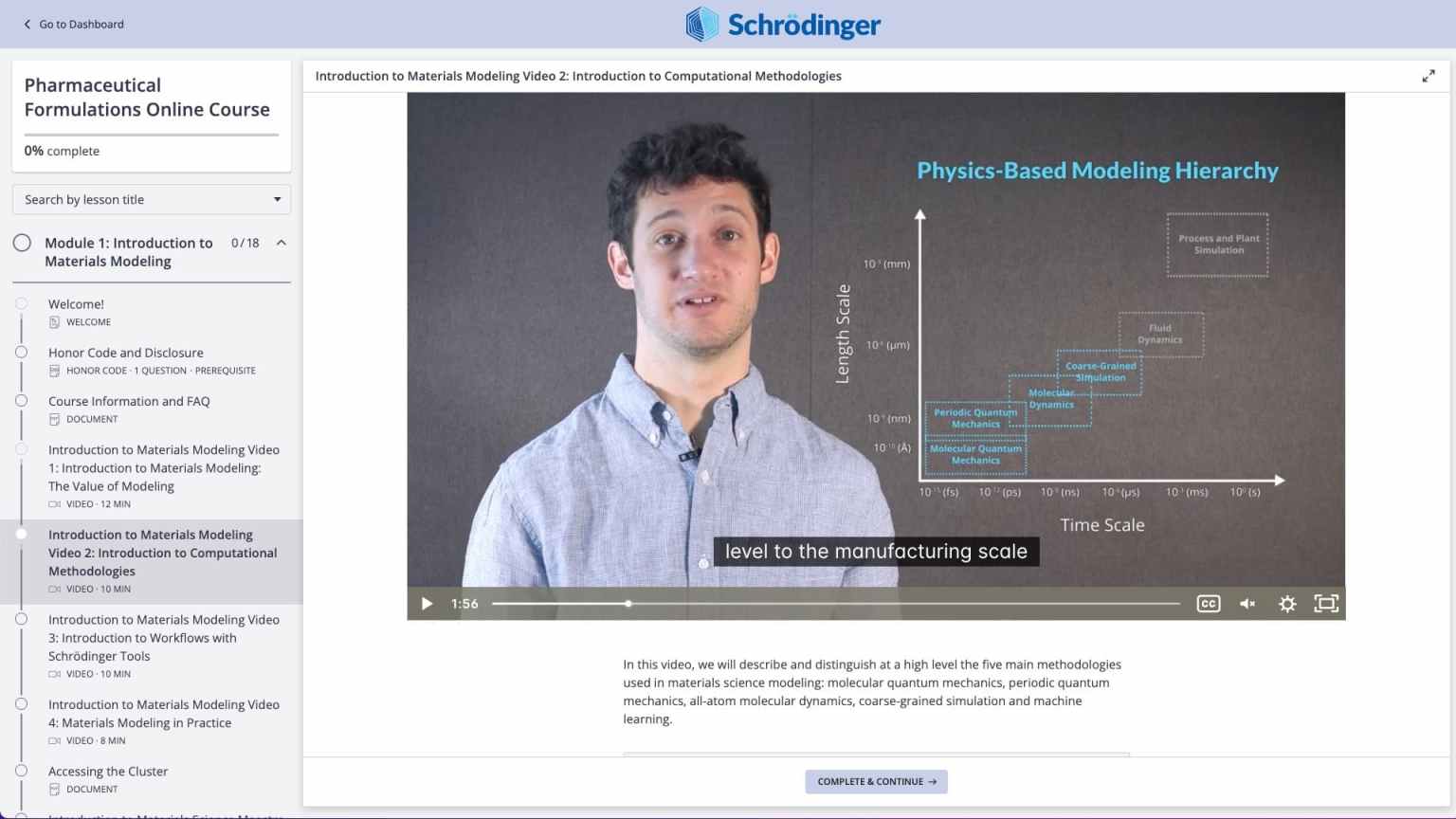
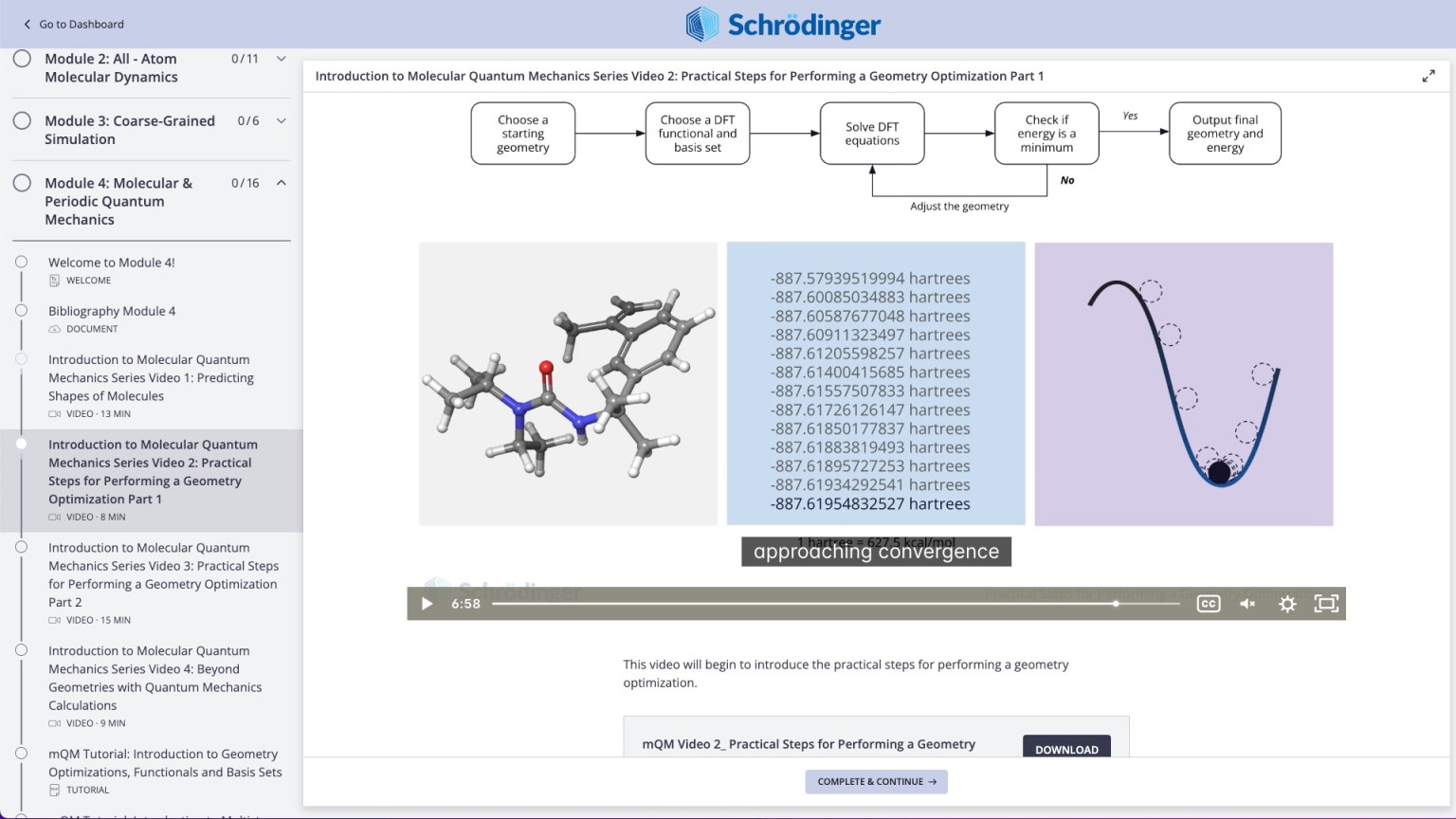
Molecular modeling for materials science applications: Battery materials course
Online certification course: Level-up your skill set in battery modeling
Learn how to apply industry-leading computational software to predict key properties of organic and organometallic compounds, determine transition state and generate reaction profiles with automated workflows and machine learning models.
- Self-paced learning content
- Hands-on access to Schrödinger software
- Guided and independent case studies
Documentation & Tutorials
Get answers to common questions and learn best practices for using Schrödinger’s software.
Key products
Learn more about the key computational technologies available to progress your research projects.
MS Formulation ML
Automated machine learning solution to generate accurate formulation-property relationships and screen new formulations with desired properties
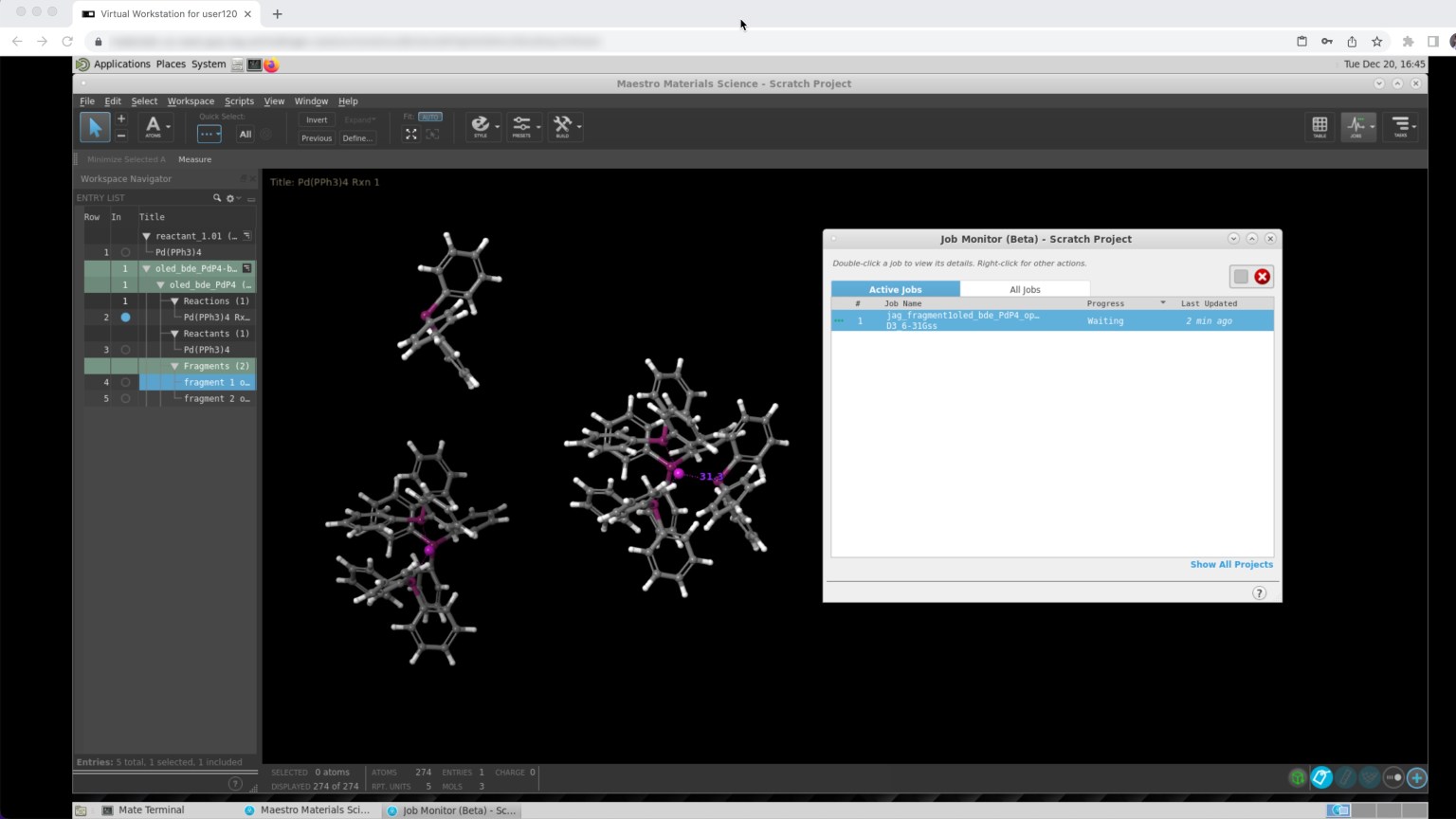
Virtual Cluster
Secure, scalable environment for running simulations on the cloud
Jaguar
Quantum mechanics solution for rapid and accurate prediction of molecular structures and properties
MS Informatics
Automated machine learning tools for materials science applications
Desmond
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
OPLS4 & OPLS5 Force Field
A modern, comprehensive force field for accurate molecular simulations
MS Transport
Efficient molecular dynamics (MD) simulation tool for predicting liquid viscosity, conductivity and diffusions of atoms and molecules
DeepAutoQSAR
Automated, scalable solution for the training and application of predictive machine learning models
MS Reactive Interface Simulator
Generate physically relevant electrode-electrolyte interface morphologies for batteries
Publications
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Schedule a consultation on Schrödinger’s battery solutions.
Contact us today to explore how you can leverage advanced simulation and AI/ML for battery materials.
Don’t see your areas of interest above? Reach out so we can help.
Form submitted
Thank you, we’ll be in touch soon.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.





























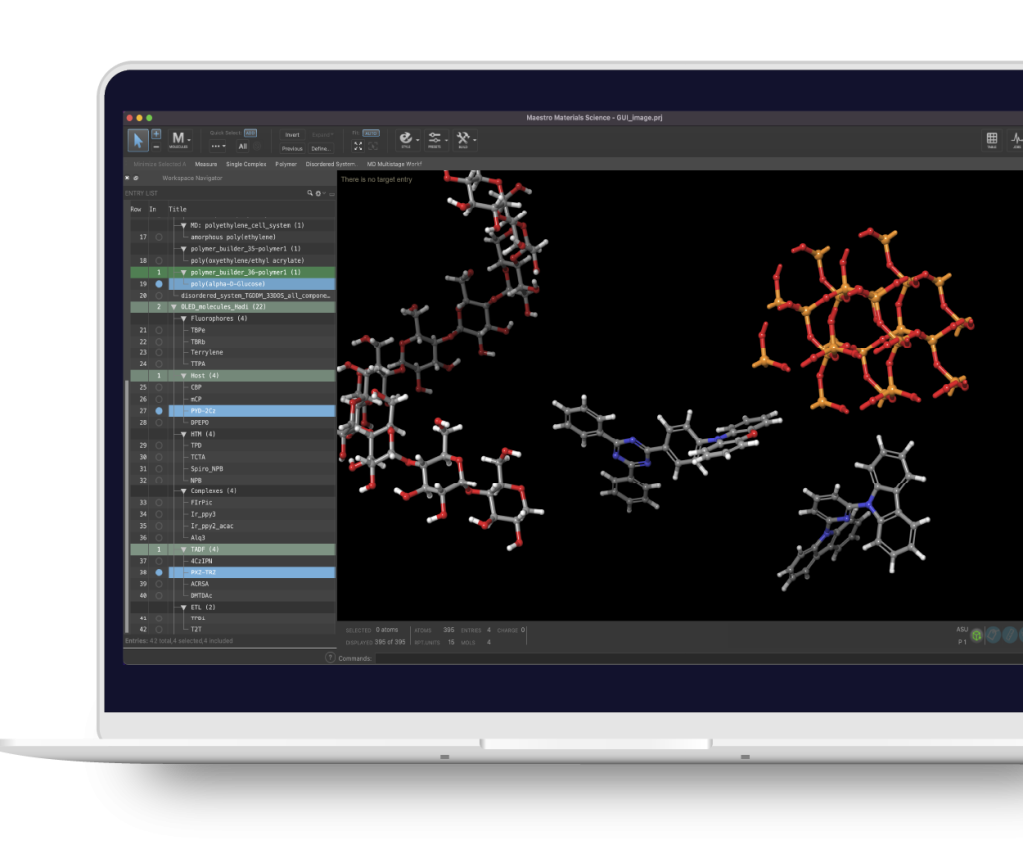
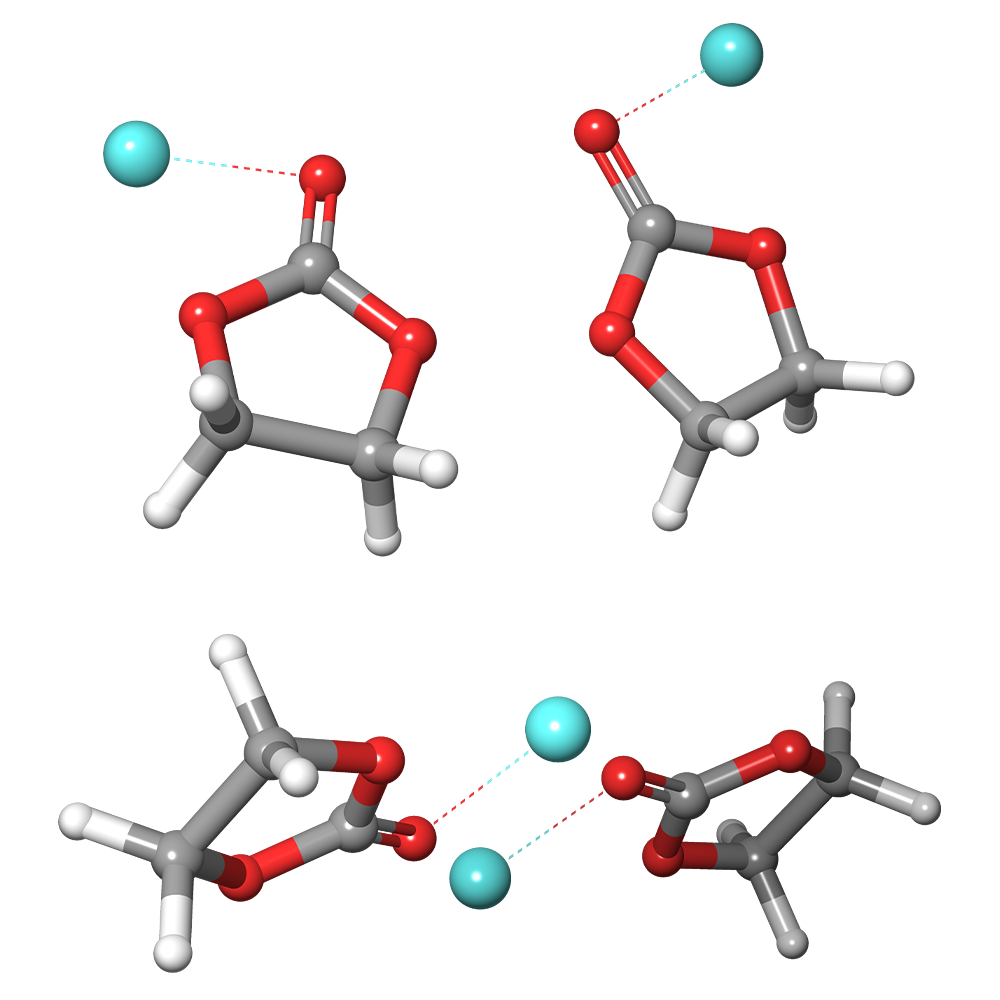


You’re in good company