Products
Augment your discovery work with a collaborative digital design ecosystem powered by industry-leading computational workflows

Augment your discovery work with a collaborative digital design ecosystem powered by industry-leading computational workflows

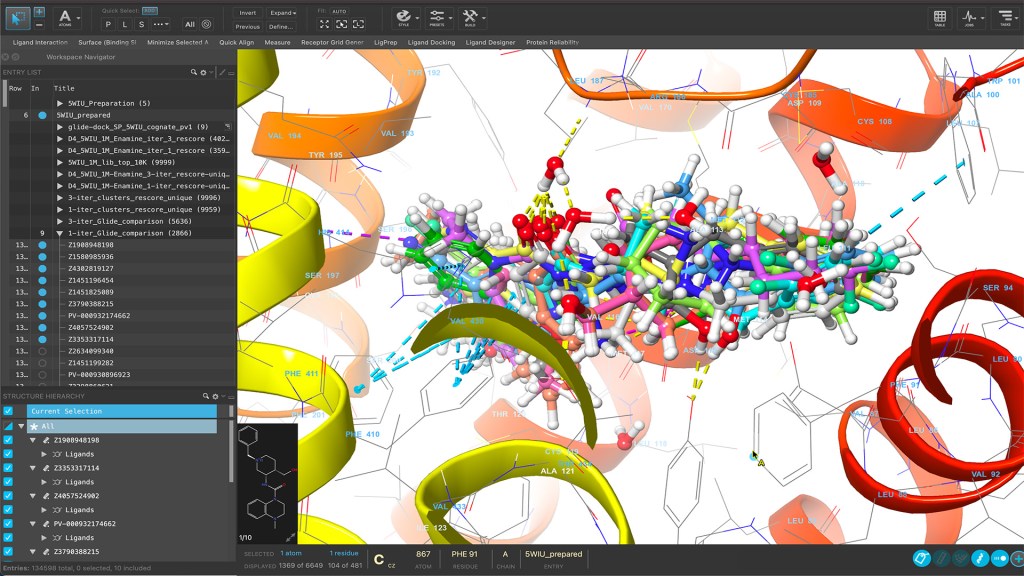
Maestro is a powerful molecular modeling environment for accessing cutting-edge physics-based molecular simulation workflows, state-of-the-art machine learning, and advanced structure visualization.
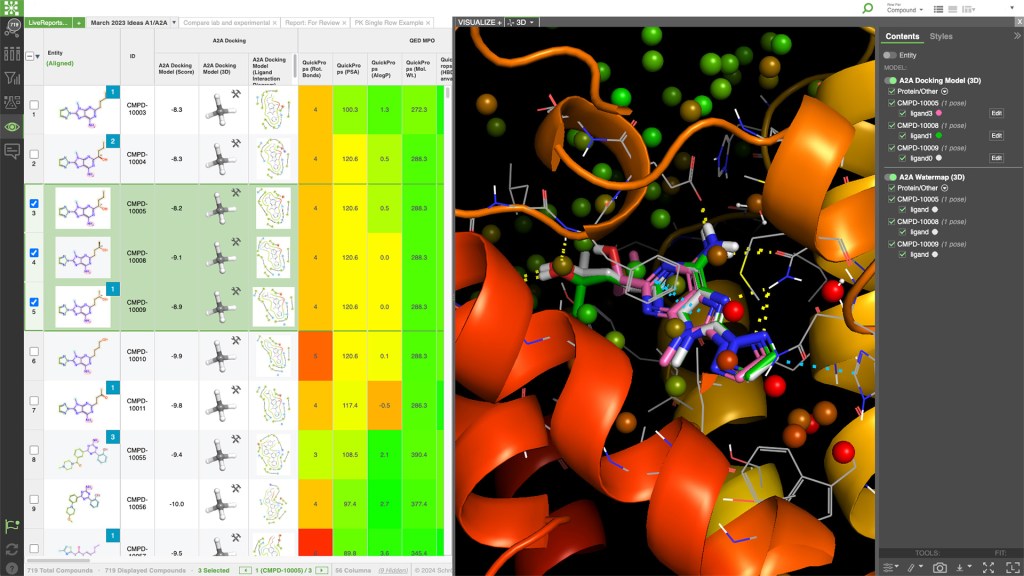
LiveDesign is a cloud-native, collaborative enterprise informatics platform with integrated 3D design, advanced cheminformatics, and data analysis workflows for the entire discovery team.
Fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement
Automated, scalable solution for the training and application of predictive machine learning models
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
Efficient tool for optimizing custom torsion parameters in OPLS4
Design solution for novel molecular materials in optoelectronic applications based on a generative algorithm
Accurate ligand binding mode prediction for novel chemical matter, for on-targets and off-targets
Quantum mechanics solution for rapid and accurate prediction of molecular structures and properties
Conformationally-dependent spectroscopic characterization based on quantum mechanics calculations
Modular, highly configurable framework for easy workflow automation and data analysis
Intuitive, interactive 3D ligand design for hit-to-lead and lead optimization
Accurate, physics-based modeling of the aqueous ionization and speciation behavior of small molecules
Physics-based solution for rapid and accurate prediction of passive membrane permeability
Services for target enablement, hit discovery, ADMET liabilities, and crystal structure prediction
Efficient coarse-grained (CG) molecular dynamics (MD) simulations for large systems over long time scales
Automatic workflow to calculate dielectric properties and refractive index
Efficient machine learning model builder for materials science applications
Atomistic simulation and analysis of charge mobility in solid-state films of organic semiconductors
Molecular dynamics (MD) modeling for predicting water loading and small molecule gas adsorption capacity of a condensed system
Automatic workflow for accurate prediction of reactivity and catalysis
Efficient molecular dynamics (MD) simulation tool for predicting liquid viscosity and diffusions of atoms and molecules
A modern, comprehensive force field for accurate molecular simulations
An easy-to-use pharmacophore modeling solution for ligand- and structure-based drug design
PyMOL is a user-sponsored molecular visualization system on an open-source foundation, maintained and distributed by Schrödinger
Integrated graphical user interface for nanoscale quantum mechanical simulations
Efficient ligand-based virtual screening of millions to billions of molecules
Virtual, novel hits from a billion-compound library delivered in one week
State-of-the-art, structure-based method for assessing the energetics of water solvating ligand binding sites for ligand optimization
Advanced docking program that leverages explicit water information in the binding site to provide more accurate scoring of ligands
Learn how to integrate Schrödinger technology into your research with molecular modeling training resources, curated by Schrödinger Education experts.
Level-up your research with molecular modeling training resources, curated by Schrödinger Education experts.