Advancing the design and optimization of drug formulations with coarse-grained molecular simulations
Scientists from AbbVie and Schrödinger gain a deep understanding of the mechanisms behind amorphous solid dispersion (ASD) dissolution behavior at the molecular level.
Executive Summary
- Evaluated dissolution profiles of different drug and polymer combinations at specified conditions
- Identified interactions that are responsible for delayed release in certain formulations
- Aligned with and complemented experimental data with visual and numeric insights at the molecular level
- Gained insights into new excipients for drug formulations to achieve targeted dissolution behavior
Challenges
Formulating small molecule drugs with low aqueous solubility in a hydrophilic polymer matrix, also known as amorphous solid dispersion (ASD), is one of the most common approaches to achieve effective drug delivery and, thus, bioavailability. Producing a high-performance ASD depends on various factors, such as the physical stability of the drug-excipient matrix, its interaction with polymers during dissolution, and the rate of drug release in an aqueous medium. Often, researchers perform numerous design and experimental iterations to achieve this goal. While hypotheses about drug release behaviors may be drawn from experimental data, a comprehensive understanding of the fundamental mechanisms and insights into molecular-level occurrences remains elusive. It’s challenging to obtain detailed drug/polymers/water interactions through experiments alone. Therefore, a more effective approach is needed to inform the selection of suitable excipients, including polymers, for specific drugs.
Approach
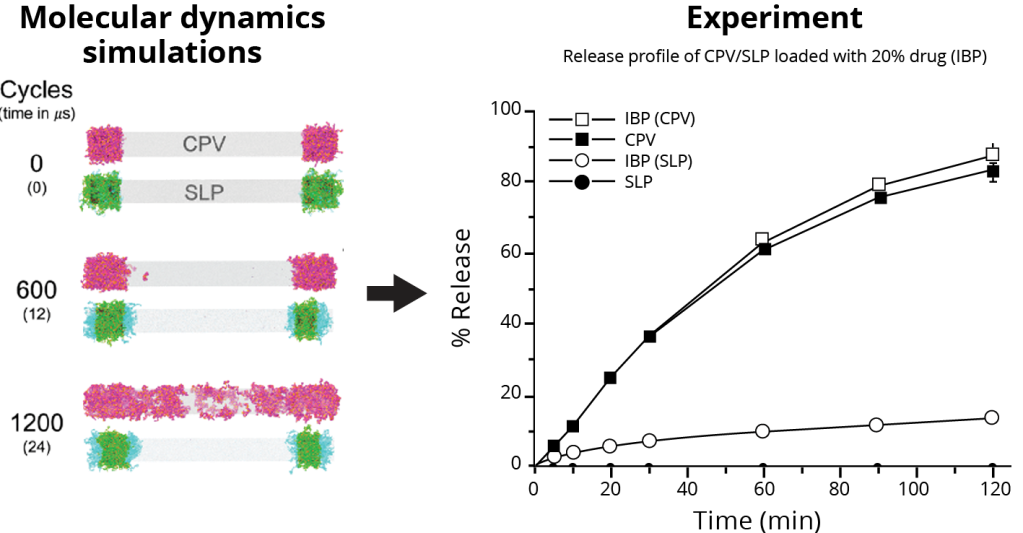
Scientists from AbbVie and Schrödinger worked together to use molecular simulations to provide insights needed to streamline time-consuming development cycles of drug formulations. A mesoscopic simulation method, dissipative particle dynamics (DPD), was employed to effectively model ASD dissolution on relatively long lengths and time scales. Two stages of the dissolution process were studied and compared with experimental investigations: the early-stage of the dissolution process, which focuses on the breakup and dissolution of the tablet at the ASD/water interface with the potential for the formation of drug-excipient particles, and the late-stage of the dissolution process where the aqueous medium contains more mature drug-excipient particles which are important for sustained supersaturation of the drug. All models and simulations were performed using the Schrödinger’s Materials Science platform and the Desmond engine for molecular dynamics (MD) and coarse-grained (CG) simulations.
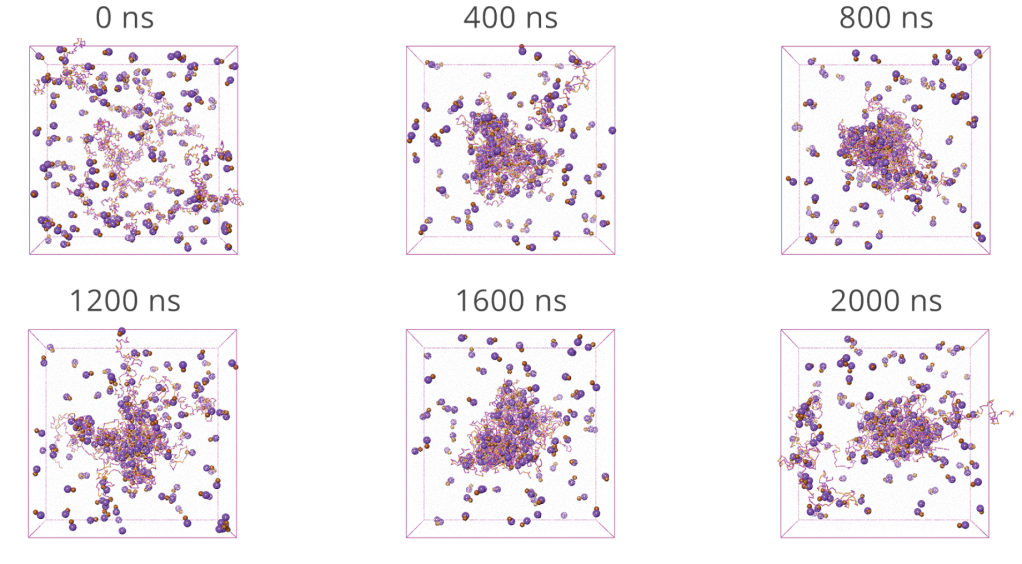
Results
- Molecular simulation results were consistent and provided visual explanations for experiments from current and previous studies:
- IBP/FFA interacts more with vinylcaprolactam in SLP than with vinylpyrrolidone in CPV
- Water interacts more with vinylpyrrolidone in CPV than with vinylcaprolactam in SLP
- Pure CPV dissolves faster than SLP, with water rapidly penetrating into CPV
- Inclusion of IBP in CPV slows down the dissolution process
- Release of IBP from SLP is slower than release from CPV
- Molecular simulations provided additional insights:
- CPV matrix showed more rapid hydration and breakup irrespective of drug presence, compared to the SLP matrix
- Surfactant-like structures at the SLP ASD−water interface slow down water penetration into the formulation and thereby the drug release
- Coherence of ASD degrades rapidly for low IBP/CPV ratios
- Water distribution within the ASDs differs significantly between the two polymers
- Within SLP, the PEG chain interacts more with water and less with drug molecules
References
-
Molecular-Level Examination of Amorphous Solid Dispersion Dissolution
Mohammad Atif Faiz Afzal, Kristin Lehmkemper, Ekaterina Sobich, Thomas F. Hughes, David J. Giesen, Teng Zhang, Caroline M. Krauter, Paul Winget, Matthias Degenhardt, Samuel O. Kyeremateng*, Andrea R. Browning, and John C. Shelley* Mol. Pharmaceutics 2021, 18(11), 3999–4014


Learn more about our collaboration with AbbVie
Advancing the design and optimization of drug formulations with combined computational and experimental approaches
Scientists from AbbVie and Schrödinger collaborated to systematically investigate
amorphous solid dispersion (ASD) dissolution behaviors by combining thermodynamic
modeling, molecular simulation, and experimental research.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.